晚期糖基化终末产物对大鼠雪旺细胞增殖和凋亡影响的体外研究
2021-02-26罗娜王琦杨文强于炎冰张黎
罗娜,王琦,杨文强,于炎冰,张黎
雪旺细胞(Schwanncells,SCs)是周围神经系统的胶质细胞,起源于神经嵴细胞,能分化成髓鞘细胞;其在髓鞘的形成和修复、维持髓鞘的厚度和绝缘性中起着重要作用[1]。此外雪旺细胞还能分泌各类神经营养因子[2],促进轴突的生长,在神经发育和再生等过程中发挥重要作用。糖尿病持续的高血糖会导致雪旺细胞的线粒体功能障碍[3],凋亡增加,导致髓鞘损伤和脱髓鞘;这可能是糖尿病周围神经病变的一个重要机制[4]。
糖尿病周围神经病(diabetic peripheral neuropathy,DPN)是糖尿病最常见的并发症,其发病机制尚未完全明确。研究显示强化血糖控制或胰腺移植并不能减少2型糖尿病DPN的发生[5-6],表明神经病变在高血糖早期已经发生。长期的高血糖导致多元醇途径激活,氧化应激和线粒体功能障碍以及晚期糖基化终末产物(advanced glycation end products,AGEs)[7]形成。过量的AGEs激活了晚期糖基化终末产物受体(receptor for advanced glycation end products,RAGE),导致细胞中活性氧(reactive oxygen species,ROS)形成,细胞氧化损伤;此外AGEs和RAGE的结合诱导氧化应激,从而导致核因子NF-κβ及其相关促炎基因的上调,引起神经功能障碍和疼痛[8-9]。雪旺细胞中RAGE过度表达与DPN的发生有关;AGEs引起关键蛋白(如胶原蛋白)、脂质和核酸的修饰有可能改变雪旺细胞的结构和功能,进而影响其包裹的轴突,导致DPN的进一步发展。因此,探讨AGEs导致雪旺细胞损伤的机制对DPN的防治有着重要意义。本研究体外培养了大鼠雪旺细胞,予以不同浓度的AGEs刺激,观察其对雪旺细胞增殖和凋亡的影响及相关蛋白表达水平的改变。
1 材料与方法
1.1 材料 RSC-96细胞(中国医学科学院基础医学研究所细胞资源中心);DMEM-H培养基,FBS,0.25%胰蛋白酶,青链霉素双抗(美国Gibco公司);CCK-8试剂盒(中国凯基公司);Caspase-3检测试剂盒,TUNEL检测试剂盒,DAPI染液(中国碧云天公司);p-Akt抗体,Akt抗体,p-GSK3β抗体,GSK3β抗体,β-Actin抗体,β-Tubulin抗体(美国CST公司);Bcl-2抗体,BAD抗体,CHOP抗体,GRP78抗体(美国Proteintech公司);兔二抗(中国碧云天公司),山羊二抗(中国普利莱公司);AGE-BSA(英国Abcam公司)。
1.2 方法
1.2.1 细胞培养及处理 将RSC-96细胞加入DMEM完全培养基(含10% FBS、1%双抗),置于37 ℃、5% CO2、95%饱和湿度培养箱中传代培养;按1∶2~1∶6传代,每2~3 d换液1次。细胞传代后,培养24 h后给予AGEs刺激,将AGEs直接溶于培养基后按照不同的浓度刺激细胞,培养一段时间后继续后续的操作。
1.2.2 细胞活力测定 将RSC-96细胞以2.0×104个/mL接种于96孔板,每孔100 μL,每组6个复孔。用DMEM完全培养基培养24 h后,更换低血清DMEM培养基(含2% FBS,1%双抗)。用0、100、250、500、1 000 μg/mL浓度AGEs处理RSC-96细胞(0 μg/mL AGEs组为对照组),刺激24 h、48 h、72 h后检测细胞的存活率。向每孔细胞加入10 μL CCK-8工作液,放回培养箱继续培养1~2 h;待培养基变成橙色后,用酶标仪检测各孔细胞在450 nm处的吸光度,即OD值。将各孔细胞的OD值减去空白OD值(培养基加CCK-8检测液,无细胞),取各孔细胞平均OD值,细胞存活率=(加药细胞OD值/对照细胞OD值)×100%。
1.2.3 Caspase-3活力测定 将RSC-96细胞以1.5×105个/mL接种于12孔板,每孔1 mL,每组4个复孔;用DMEM完全培养基培养24 h后,更换低血清DMEM培养基。用0、100和500 μg/mL浓度的AGEs处理RSC-96细胞48 h后,检测其Caspase-3活力。用胰酶将贴壁细胞消化下来,600 g、4 ℃离心5 min收集细胞,小心吸除上清,PBS清洗1次后,加入裂解液,重悬沉淀,冰浴裂解15 min。每组细胞取少量样品用Bradford法测定蛋白浓度,再取50 μL样品用于检测ρNA(ρ-nitroaniline)。每组样品中加入40 μL缓冲液和10 μL Ac-DEVD-ρNA(acetyl-Asp-Glu-Val-Asp ρ-nitroanilide),空白对照用50 μL裂解液代替蛋白样品,混匀后37 ℃孵育1~2 h,颜色明显变黄后测定样品的OD值。将各孔细胞的OD值减去空白对照的OD值,取各孔细胞平均OD值,Caspase-3活力=(实验组OD值/对照组OD值)×(对照组蛋白浓度/实验组蛋白浓度)×100%。
1.2.4 细胞凋亡检测 将RSC-96细胞以1.5×105个/mL接种于24孔板(内含细胞爬片),每孔0.5 mL,每组3个复孔。用DMEM完全培养基培养24 h后,更换低血清DMEM培养基,用0、100和500 μg/mL浓度的AGEs处理RSC-96细胞72 h后,检测细胞凋亡。贴壁细胞用PBS洗一遍后加入4%多聚甲醛固定30 min,PBS洗一遍后加入通透液(含0.3% Triton X-100的PBS)室温孵育5 min。按说明书配置TUNEL检测液,细胞用PBS洗涤两次后加入50 μL TUNEL检测液,37 ℃避光孵育60 min,PBS洗3次后加入DAPI染色液,室温避光孵育10 min。洗去DAPI染色液后用抗荧光猝灭封片液封片,在荧光显微镜下观察、拍照,用ImageJ软件分析结果;计算凋亡指数(凋亡指数=凋亡细胞数/总细胞数×100%)。
1.2.5 细胞蛋白检测 采用免疫印迹法。将RSC-96细胞以1.5×105个/mL接种于6孔板,用DMEM完全培养基培养24 h后,更换低血清DMEM培养基。用0、100和500 μg/mL浓度的AGEs处理RSC-96细胞72 h后,提取细胞总蛋白。根据蛋白浓度算出每组样品体积,将蛋白样品与蛋白上样缓冲液按5∶1混匀后100 ℃加热5 min。将蛋白样品混合物和蛋白Maker加入10% SDS-PAGE凝胶内,以150 V恒压电泳60 min。然后取出凝胶,以250 mA恒定电流湿转60 min,用含有5%脱脂奶粉的TBST溶液室温封闭1 h。用一抗稀释液稀释一抗,将PVDF膜在稀释的一抗中4 ℃孵育过夜,TBST洗涤3次,每次10 min;用二抗稀释液稀释二抗,将PVDF膜在稀释的二抗中室温孵育1 h,TBST洗涤3次,每次10 min;将ECL显影液按A液与B液1∶1配好后,将膜在显影液中避光孵育30~60 s,然后放入成像系统,曝光成像。用Image J软件分析目标条带的灰度值,以内参的灰度值作为内部对照。
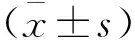
2 结 果
2.1 AGEs对RSC-96细胞增殖的影响 与对照组细胞相比,培养24 h、48 h时各浓度AGEs组RSC-96细胞的活力无显著下降;但培养72 h时,各浓度AGEs组RSC-96细胞的活力均明显降低(均P<0.05)(图1);表明AGEs处理72 h后RSC-96细胞的增殖能力减弱。
与Control组比较*P<0.05图1 不同浓度AGEs组RSC-96细胞培养24~72 h相对活力变化
2.2 AGEs对RSC-96细胞凋亡的影响
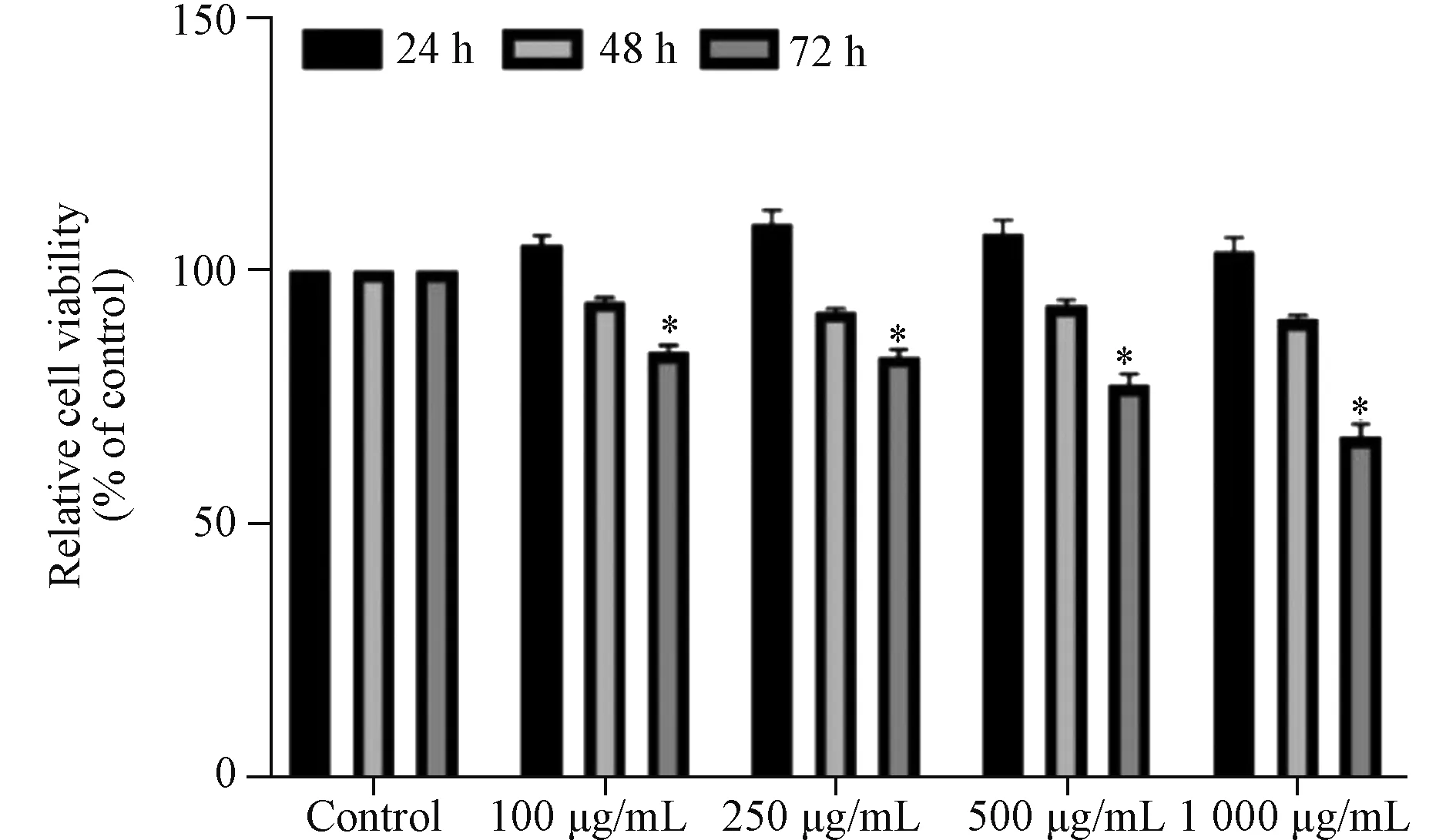
2.2.1 AGEs组与对照组RSC-96细胞Caspase-3活性比较 与对照组相比,100 μg/mL和500 μg/mL AGEs组RSC-96细胞的Caspase-3活性均明显增强(均P<0.05)。见图2。
与Control组比较*P<0.05图2 不同浓度AGEs组与对照组RSC-96细胞Caspase-3活性比较
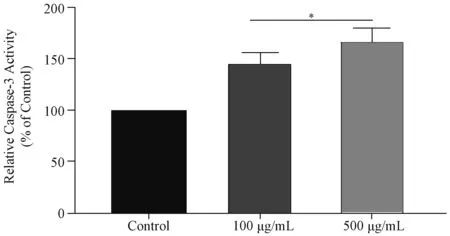
2.2.2 AGEs组与对照组RSC-96细胞凋亡指数比较 随着AGEs浓度的增加,凋亡细胞(绿色荧光标记阳性细胞)数增多(图3)。与对照组相比,100 μg/mL和500 μg/mL AGEs组RSC-96细胞的凋亡指数均明显增高(均P<0.05)(图4)。
2.3 AGEs对RSC-96细胞Akt通路的影响 与对照组相比,100 μg/mL和500 μg/mL AGEs组RSC-96细胞的P-Akt、p-GSK3β蛋白水平明显降低;p-Akt/Akt和p-GSK3β/GSK3β明显降低。表明其Akt和GSK3β磷酸化受到了抑制,AGEs可以抑制RSC-96细胞Akt通路的激活。见图5-7。
2.4 AGEs对RSC-96细胞凋亡蛋白表达的影响 与对照组相比,100 μg/mL和500 μg/mL AGEs组RSC-96细胞的BAD蛋白表达水平升高,而Bcl-2蛋白表达水平降低(图8);表明AGEs可以促进RSC-96细胞促凋亡蛋白的表达,抑制RSC-96细胞抑凋亡蛋白的表达。
2.5 AGEs对RSC-96细胞内质网应激的影响 与对照组相比,100 μg/mL和500 μg/mL AGEs组RSC-96细胞的CHOP蛋白表达水平升高,而GRP78蛋白表达水平降低(图9);表明AGEs导致了RSC-96细胞的内质网应激增强且处于内质网应激晚期失代偿。
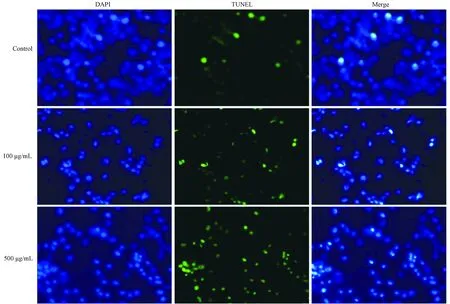
图3 各组RSC-96细胞凋亡检测结果(荧光染色,×100)

与Control组比较**P<0.05图4 不同浓度AGEs组与对照组RSC-96细胞凋亡指数比较图5 各组RSC-96细胞Akt通路蛋白免疫印迹检测
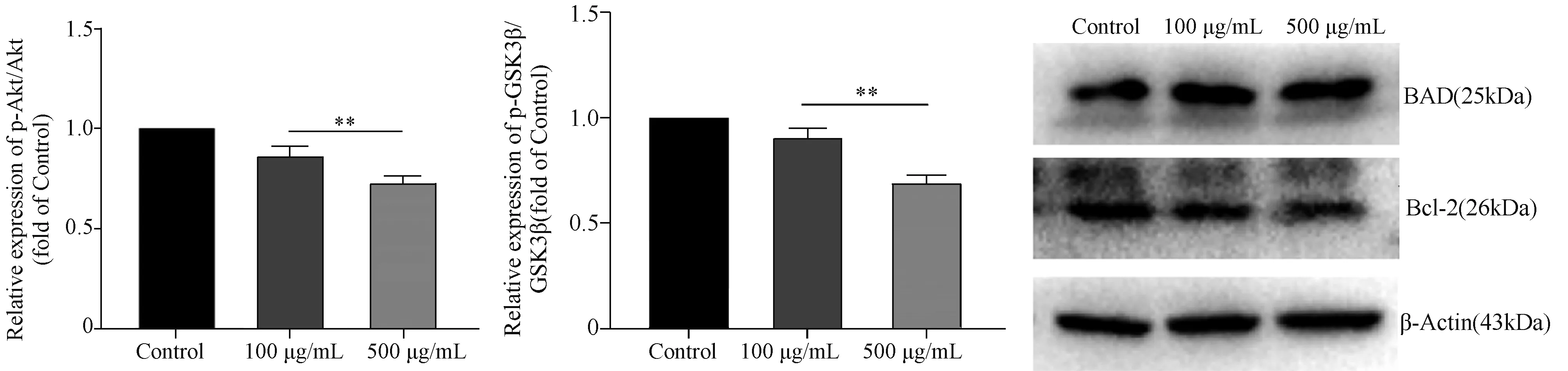
与Control组比较**P<0.05图6 不同浓度AGEs组与对照组RSC-96细胞p-Akt/Akt蛋白水平比较与Control组比较**P<0.05图7 不同浓度AGEs组与对照组RSC-96细胞p-GSK3β/GSK3β比较图8 各组RSC-96细胞的BAD、Bcl-2蛋白表达水平
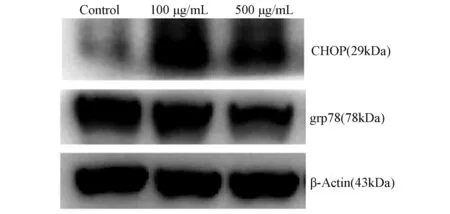
图9 各组RSC-96细胞的CHOP、GRP78蛋白表达水平
3 讨 论
中国糖尿病患者呈快速增长趋势,糖尿病前期患者的比例也很高,超过半数的糖尿病患者会发展成为DPN,严重影响患者生活质量[10]。临床上对DPN的治疗方法是控制血糖和疼痛的对症治疗,尚无有效的药物阻止DPN的发生。DPN发病机制十分复杂,尚未完全阐明。目前认为其与遗传易感性、胰岛素抵抗、高血糖、低度炎症状态、血管内皮细胞功能紊乱和凝血功能异常等多种因素有关。高血糖可导致多元醇途径激活、AGEs形成增加、蛋白激酶C途径激活以及己胺糖通路激活等。高血糖时线粒体电子传递链过氧化物产生过量引起氧化应激是以上各种途径的共同机制。因此,研究DPN的病理机制对于寻找延缓DPN进展的关键药物意义重大。
高血糖导致过量的AGEs累积是DPN的重要发病机制,AGEs的形成和累积与糖尿病的进展呈正相关。研究发现,AGEs在糖尿病患者和实验动物的周围神经中积累,而抗糖基化药物可以抑制AGEs的形成,改善实验性糖尿病大鼠的神经病变[11-12]。AGEs由赖氨酸侧链和大分子(氨基酸、蛋白质、磷脂和核酸)末端氨基还原碳水化合物的非酶反应(糖基化反应)生成,这个过程开始形成可逆的希夫碱,然后得到更稳定的早期糖基化产物,经过进一步的复杂反应,如重排、脱水和缩合,生成了不可逆的交联荧光蛋白衍生物,称为AGEs[13]。机体组织中AGEs累积会对蛋白质、脂类和DNA的功能产生不利影响,诱发炎症和活性氧浓度的升高,最终影响细胞代谢活动;这些因素进一步导致AGEs累积,引起机体疾病如糖尿病、炎症、心血管疾病和神经退行性疾病[8,14-15]。在血管壁中AGEs可激活细胞内信号转导通路,从而引起生长因子、细胞粘附分子及细胞因子表达增加,抑制细胞复制并诱导细胞凋亡[16]。然而,AGEs在DPN中的作用机制尚不清楚。
AGEs有许多不同的来源,葡萄糖通过糖基化、自氧化和其他代谢途径形成免疫化学性质不同的AGEs。由早期糖基化产物(amadori product)分解产生的、甘油醛(糖酵解中间产物)产生的、乙醇醛(希夫碱分解产物)产生的、甲基乙二醛(甘油醛和糖基化产物分解产物)产生的和葡萄糖自氧化产生的AGEs分别命名为AGE-1~5[17]。这5种AGEs抗体均能在糖尿病患者的血液中检测到[18]。这些不同的内源性AGEs对神经元、视网膜细胞和肾细胞有不同的生物学效应[19];AGE-2(甘油醛衍生的AGEs)能促进神经元的凋亡[17];AGE-2和AGE-3(乙醇醛衍生的AGEs)能显著抑制SCs的复制,诱导其凋亡;而AGE-1(葡萄糖衍生的AGEs)对SCs的增殖和凋亡无明显影响[20]。研究还发现AGE-2和AGE-3能促进 SCs核因子NF-κβ的活化以及TNF-α和IL-1β的分泌,还能导致细胞氧化应激增强和线粒体功能障碍。本研究采用Abcam公司的AGEs,由BSA与乙醇醛发生反应得到的AGE-BSA进行细胞实验;结果表明AGEs以剂量和时间依赖性方式影响RSC-96细胞的存活,增强细胞Caspase-3活性,导致细胞的凋亡数明显增加。由此推测AGEs直接导致了雪旺细胞的凋亡,进而引起神经脱髓鞘和周围神经再生受损;这可能是高血糖引起周围神经病变的重要机制。
Akt是细胞存活的重要调节因子,可以抑制细胞凋亡。Akt激活后会磷酸化其下游的调节蛋白,最著名的是GSK3,还包括雷帕霉素(mTOR)、TSC2、caspase-9和PRAS40 (AKT1S1)。Akt在促进细胞增殖、分化、凋亡、血管生成和代谢等方面具有相对广泛的下游效应[21],参与葡萄糖代谢和储存的糖原合酶受GSK3和其他几种下游蛋白的调控;此外,mTORC1通过固醇反应元件结合蛋白(SREBP)转录因子调节脂质代谢;这些因素通过防止蛋白质分解来促进细胞生长,通过激活核苷酸合成来刺激细胞增殖,并在不同情况下调节细胞自噬[22]。因此可以推断,Akt信号传导途径在糖尿病及其相关并发症、癌症的发生和进展、损伤、修复和重塑等过程中起到至关重要的作用。研究发现Akt在周围神经系统轴突包裹和髓鞘厚度调节中起关键作用[23];糖尿病及其并发症的发展受到Akt信号通路的调节[24-25]。动物实验研究发现,DPN大鼠的背根神经节和坐骨神经中Akt的激活受到了抑制[26-27];在细胞实验中,药物激活Akt信号通路可以保护细胞免受高糖和氧化应激等造成的细胞损伤[28-29];这给寻找神经营养药物一些提示。相反,抑制Akt的激活可以导致细胞的凋亡[30],这可能成为癌症治疗的一个靶点。本研究观察到AGEs处理的RSC-96细胞,磷酸化的Akt-Sre473和磷酸化的GSK3β-Ser21/9的表达水平降低,表明Akt信号通路受到了抑制。
Bcl-2和BAD是Akt/GSK3β信号通路的下游调节蛋白,Bcl-2可抑制细胞凋亡,BAD则促进细胞凋亡[25]。本研究中, AGEs处理的RSC-96细胞中Bcl-2表达水平降低,而BAD表达水平增高;提示AGEs可能通过阻断Akt/GSK3β信号通路,影响其下游凋亡相关蛋白的表达来诱导凋亡。在内质网应激的3种主要信号通路中,PERK (protein kinase RNA-like ER kinase)/CHOP(CCAT-enhancer-binding protein homologous protein)通路被认为与细胞凋亡密切相关。内质网应激的早期阶段是一种旨在维持内质网稳态的适应性反应,被称为未折叠蛋白反应(unfolded protein reaction,UPR),由葡萄糖调节蛋白78(glucose regulatory protein 78,GRP78)和内质网伴侣蛋白调控;PERK信号通路发生在内质网应激早期,通过抑制蛋白质的合成对细胞起保护作用,促进细胞的生存;如内质网应激长期存在,可诱导细胞凋亡的内在途径[31];PERK通过诱导CHOP的表达而促进细胞凋亡,CHOP可抑制Bcl-2的表达,增加活化Caspase-3的表达水平,导致细胞死亡。GRP78在内质网应激早期表达增多,而在晚期则表达减少。在本研究中,AGEs刺激后的RSC-96细胞GRP78蛋白水平降低,而CHOP蛋白水平升高,Caspase-3的表达水平增加;表明AGEs造成了RSC-96细胞的内质网应激,且处于内质网应激晚期。
有研究认为Akt激活可以减轻内质网应激[32],Akt失活则导致内质网应激[33]。也有研究发现,CHOP抑制Akt的激活,而GRP78则增强Akt的激活,促进细胞存活[34-35]。本研究推测Akt信号通路和内质网应激通路相互作用,互相影响。通过激活Akt、抑制内质网应激可以促进细胞的存活,这为在高血糖和氧化应激状态下保护神经、减少雪旺细胞凋亡提供了很好的方法。本研究通过AGEs刺激大鼠雪旺细胞实验发现了DPN可能的机制,即AGEs通过抑制Akt的激活,增强内质网应激导致雪旺细胞的凋亡;结果提示可以通过寻找激活Akt或抑制内质网应激的药物减缓DPN的进展。
