Rapid synthesis and characterization of bridged (bis-, tri- and tetra-) aryl carboxylic acid derivatives at room temperature by ultrasonic irradiation
2020-12-22CHENLianqingYANGGengtaoWUChao
CHEN Lianqing, YANG Gengtao, WU Chao
(Hubei Key Laboratory of Catalysis and Materials Science,College of Chemistry and Materials Science,South-Central University for Nationalities, Wuhan 430074, China)
Abstract Ultrasonic radiation was applied to perform the esterification and acylation reaction of alcohols and amines with acid chlorides. 28 bridged (bis-, tri- and tetra-) aryl carboxyl derivatives were synthesized at room temperature, with the structures characterized by IR, 1H NMR, MS, elemental analysis and so forth. The results indicated that the reactions were accomplished with high yield in short reaction time under mild conditions, and with no use of catalyst. This green approach represents a new general route for the synthesis of aryl carboxylic acid derivatives.
Keywords ultrasonic radiation; bridged (bis-, tri- and tetra-) aryl carboxylic acid derivatives
Carboxylic acid is one of the most important intermediates in organic chemistry because it can be transformed into esters, acyl halides, amides, anhydride and other derivatives. It has attracted much attention due to its importance in biology, organic chemistry, natural products, agriculture, pharmaceuticals and so on[1]. In these research areas, esters and amides are the focus of research. The ester compounds are very important fine chemicals, pharmaceutical intermediates, synthesis material intermediates, etc. For instance, phosphotriester compounds were used to produce phosphoric acid, which could be used as flame retardant; pentaerythritol tetraester is an environmentally friendly lubricant due to its comprehensive properties[2]. The amines are also one of the most common compounds in organic chemistry, and have important applications in the fields of drug chemistry, biochemistry and polymer synthesis[3]. The amines, including bisamides, triamides and tetraamides, have a wide range of properties and can be used for anti-inflammatory, antibacterial, anti-tumor, herbicidal, and insecticidal applications. Among them,N,N′-alkylene diamide is the key structural subunits of pseudopeptide framework[4], and 1, 3, 5-benzenetricaboxylic acid is a nucleating agent which can effectively improve the performance of the polypropylene[5].
The traditional synthesis of carboxylic ester compounds involves heating or refluxing for a long time under the action of catalyst. For example, monoester is generally prepared by using concentrated sulfuric acid to catalyze the dehydration of alcohol and acid. However, reports about the synthesis of bridged (bis-,tri- andtetra-) aryl compounds are rare. At present, there are mainly esterification method, transesterification method and heating/refluxing two-step reaction for the synthesis of ester compounds, such as the direct esterification of carboxylic acid and perfluoroalcohol mediated by XtalFluor-E[6]. But this synthetic route has many disadvantages, such as long reaction time, many side reactions and complex post-treatment. Synthesis of five-membered cyclic carbonate by thermal transesterification has also been reported by GUIDI et al[7]. However, this method needs multi-step heating, complicated operation and high energy consumption. PENG[8]synthesized methyl cyanoacetate by reflux heating of cyanoacetic acid and methanol with sulfuric acid as catalyst, and then obtained the corresponding ester by cyano alcoholysis.
The formation of the amine bond is very important in organic reactions. It is found in proteins and peptides, a large number of drugs, natural products, fine chemicals and lubricants. The synthesis of amide compounds is commonly used in acid halogen reaction, mixed anhydride reaction and condensation reaction. DE SOUZA used NaClO for the oxidative coupling of amine, which required heating for 12 h and the process was complicated[9]. The RuFG catalyst prepared by Ramen Jamatia was used in water medium to convert an aromatic aldehyde into the corresponding primary amine[10]. JIN synthesizedN′-(2-hydroxy-4-methyl-benzoylamide)-2-imino-4-thiazolidinone with an amine group[11]. If such methods were to be used in the synthesis ofbis-,tri- andtetra-amides, harsh reaction conditions such as highly active acylating agents, high temperature, and high pressure would still be required, which was not conducive to practical production. Although esters and amides can be synthesized by some particular methods, none of them represents a general route.
The ultrasonic chemistry is a recently developed area, which uses cavitation effect to break the specific chemical bond and accelerate the chemical reaction[12]. In recent years, the ultrasonication technology has been widely used in many fields, such as chemistry, medicine, petrochemicals, etc. The promoting effect of ultrasonic wave is due to the cavitation effect and secondary effect when ultrasonic wave propagates in the medium. By utilizing these two effects, reactions that could not take place under normal conditions may occur smoothly and new reaction channels may be opened up.
1 Experimental
1.1 Reagents and apparatus
Alcohol,n-propyl alcohol,n-butyl alcohol,benzilalcohol,phenol,2-alcohol thiophene,cyclopentanol,isophthaloyl chloride,1,3,5-benzenetricarboxylic acid,trimesoyl chloride,1,2,4,5-benzenetetracarboxylic acid,1,4,5,8-naphthalenetetracarboxylic acid,aniline,benzylamine,[1,1′-biphenyl]-3,3′,4,4′-tetracarboxylic acid,p-aminotoluene,p-fluoroaniline,n-butylamine,2-aminothiophene,cyclopentylamine,propylamine,dichloromethane,sodium hydrate,ethyl acetate.
Ultrasonic cleaning instrument (KQ3200E,150 W); Fourier transform infrared spectrometer(NEXUS470,Nicolet); rotary evaporator (RE-52, Shanghai Yilong Bio-chemical Plant); circulating water vacuum pump (SHZ-D III, Wuhan Kohl); all-digital nuclear magnetic resonance spectrometer (AVANCE III,400 MHz, Switzerland Brooke), dichloromethane as solvent, TMS as internal standard; digital melting point meter (WRS-1B, Shanghai Jingke); element analyzer (Vario-EL III CHNS, USA).
1.2 Preparation
1.2.1 Preparation step of esterification reaction
Take the synthesis ofE2ahas an example: add 10.5 mmol of ethanol and 10.5 mmol of triethylamine to a 100 mL flask, dissolve in 20 mL of methylene chloride, and then add 10 mL of a solution of methylene chloride with 5 mmol of chlorine of terephthalic acid. Put the flask in the ultrasonic washing instrument to react 20 min. The crude product was extracted with 100 mL ethyl acetate and 30 mL water. The solvent was evaporated, and finally dried in vacuum. The other esters were synthesized according to a similar method.
1.2.2 Preparation step of acylation reaction
Take the synthesis ofA2hpas an example(Fig.1): add 10.5 mmol of aniline to a 100 mL flask, dissolve in 40 mL of methylene chloride, and then add 10 mL of a solution of methylene chloride with 5 mmol of chlorine of terephthalic acid. Put the flask in the ultrasonic washing instrument to react 15 min. After filtration, the filter cake was washed for many times with 20 mL 10% NaOH solution to remove acid residue. A white solid product was finally obtained. Other amine derivatives were synthesized in a similar way.

Fig.1 Synthesis route of aryl carboxylic acid derivatives图1 芳基羧酸衍生物合成路线
2 Results
2.1 Ester with the number of groups n=2
The synthetic route of esterification is shown in Fig.2.
ProductE2ah: white solid, 99% yield. m.p. 75-76 ℃.1H NMR (CDCl3, 400 MHz)δ: 8.10 (s, 4H, Ar-H), 4.41(m, 4H, C-H), 1.42 (t, 6H, C-H); IR (KBr,ν/cm-1): 2990, 2973, 1718, 1518, 1410, 1277, 1247, 729. Anal. calc for C12H14O4: C 64.85, H 6.35; found C 64.89, H 6.32. MS (FAB):m/e, 222 (M+).
ProductE2bh: white solid, 83% yield. m.p. 97-98 ℃.1H NMR (CDCl3, 400 MHz)δ: 8.03 (s, 4H, Ar-H), 4.23 (t, 4H, C-H), 1.73 (m, 4H, C-H), 0.96 (t, 6H, C-H); IR (KBr,ν/cm-1): 2969, 2881, 1722, 1578, 1465, 1272, 1204, 731. Anal. calc for C14H18O4: C 67.18, H 7.25; found C 67.23, H 7.17. MS (FAB):m/e, 250 (M+).
ProductE2ch: white solid, 92% yield. m.p. 120-121 ℃.1H NMR(CDCl3, 400 MHz)δ: 8.02 (s, 4H, Ar-H), 4.28 (t, 4H, C-H), 1.70 (m, 4H, C-H), 1.42 (m, 4H, C-H), 0.91 (t, 6H, C-H); IR (KBr,ν/cm-1): 2961, 2874, 1724, 1578, 1465, 1273, 1104, 731. Anal. calc for C16H22O4:C 69.04, H 7.97; found C 69.11, H 7.93. MS (FAB):m/e, 278 (M+).
ProductE2dh: white solid,95% yield. m.p. 240-241 ℃.1H NMR (CDCl3, 400 MHz)δ: 8.06 (s, 4H, Ar-H), 7.33 (m, 10H, Ar-H), 5.31(s, 4H, C-H); IR (KBr,ν/cm-1): 3062, 2936, 1715, 1500, 1453, 1406, 1378, 1275, 1128, 736. Anal. calc for C22H18O4: C 76.29, H 5.24;found C 76.32, H 5.26. MS (FAB):m/e, 346 (M+).
ProductE2ai: white solid, 99% yield.m.p.73-74 ℃.1H NMR (CDCl3, 400 MHz)δ: 8.67 (s, 1H, Ar-H), 8.20 (d, 2H, Ar-H), 7.51 (t, 1H, Ar-H), 4.41(m, 4H, C-H), 1.40 (t, 6H, C-H); IR (KBr,ν/cm-1): 2983, 1304, 1240, 730. Anal. calc for C12H14O4: C 64.85, H 6.35; found C 64.88, H 6.31. MS (FAB):m/e, 222 (M+).
ProductE2bi: white solid, 96% yield.m.p.95-96 ℃.1H NMR (CDCl3, 400 MHz)δ: 8.68 (s, 1H, Ar-H), 8.21 (d, 2H, Ar-H), 7.52 (t, 1H, Ar-H), 4.30 (t, 4H, C-H), 1.80 (m, 4H, C-H), 1.03 (t, 6H, C-H); IR (KBr,ν/cm-1): 2969, 1722, 1390, 1300, 1237, 729. Anal. calc for C14H18O4: C 67.18, H 7.25; found C 67.12, H 7.29. MS (FAB):m/e, 250 (M+).
ProductE2ci: white solid, 88% yield. m.p.117-118 ℃.1H NMR (CDCl3, 400 MHz)δ: 8.60 (s, 1H, Ar-H), 8.15 (d, 2H, Ar-H), 7.45 (t, 1H, Ar-H), 4.28 (t, 4H, C-H), 1.69 (m, 4H, C-H), 1.40 (m, 4H, C-H), 0.91(t, 6H, C-H); IR (KBr,ν/cm-1): 2961, 2874, 1723, 1609, 1465, 1305, 1241, 730. Anal. calc for C16H22O4: C 69.04, H 7.97; found C 69.07, H 7.73. MS (FAB):m/e, 278 (M+).
ProductE2di: white solid, 85% yield.m.p.228-229 ℃.1H NMR (CDCl3, 400 MHz)δ: 8.64 (s, 1H, Ar-H), 8.20 (d, 2H, Ar-H), 7.34 (m, 10H, Ar-H), 5.32 (s, 4H, C-H); IR (KBr,ν/cm-1): 3033, 2956, 1716, 1237, 1128, 757, 700. Anal. calc for C22H18O4: C 76.29, H 5.24; found C 76.33, H 5.28. MS (FAB):m/e, 346 (M+).
2.2 Ester with the number of groups n=3
The synthetic route of esterification is shown in Fig.2.
ProductE3cj: white solid, 89% yield.m.p.231-232 ℃.1H NMR (CDCl3, 400 MHz)δ: 8.25 (d, 2H, Ar-H), 7.42 (t, 1H, Ar-H), 4.28 (m, 4H, C-H), 2.76 (m, 5H, C-H), 1.03 (t, 9H, C-H), 0.83 (m, 9H, C-H); IR (KBr,ν/cm-1): 3028, 1643, 1544, 1468, 1305, 1241, 817. Anal. calc for C21H30O6: C 66.65, H 7.99; found C 66.59, H 7.93. MS (FAB):m/e, 378 (M+).
ProductE3ej: white solid, 90% yield.m.p.258-259 ℃.1H NMR (CDCl3, DMSO, 400 MHz)δ: 8.83 (s, 3H, Ar-H), 8.48 (d, 2H, Ar-H), 7.81 (m, 5H, Ar-H), 7.18 (d, 4H, Ar-H), 6.98 (d,4H, Ar-H); IR (KBr,ν/cm-1): 3033, 1665, 1562, 1451, 1310, 1245, 816. Anal. calc for C27H18O6: C 73.97, H 4.14; found C 73.91, H 4.18. MS (FAB):m/e, 438 (M+).
ProductE3fj: white solid, 92% yield.m.p.276-277 ℃.1H NMR (CDCl3, 400 MHz)δ: 8.87 (d, 2H, Ar-H), 8.35 (d, 3H, Ar-H), 7.68 (m, 4H, Ar-H), 6.83 (m, 3H, Ar-H); IR (KBr,ν/cm-1): 3028, 1669, 1571, 1436, 1307, 1241, 811. Anal. calc for C21H12O6S3: C 55.25, H 2.65; found C 55.26, H 2.61. MS (FAB):m/e, 455 (M+).
2.3 Ester with the number of groups n= 4
The synthetic route of esterification is shown in Fig.2.
ProductE4gk: white solid, 87% yield.m.p.286-287 ℃.1H NMR(CDCl3, 400 MHz)δ: 8.72 (s, 2H, Ar-H), 6.89 (s, 3H, C-H), 3.28 (d, 4H, C-H), 2.19 (m, 11H, C-H), 1.84 (m, 9H, C-H), 1.57 (m, 12H, C-H); IR (KBr,ν/cm-1): 3010, 1674, 1556, 1443, 1307, 1245, 812. Anal. calc for C30H38O8: C 68.42, H 7.27; found C 68.49, H 7.10. MS (FAB):m/e, 526 (M+).
ProductE4el: white solid, 74% yield.m.p.294-295 ℃.1H NMR(CDCl3, 400 MHz)δ: 8.23 (s, 2H, Ar-H), 8.11 (d, 2H, Ar-H), 7.79 (s, 6H, Ar-H), 7.46 (t, 8H, Ar-H), 7.14 (m, 6H, Ar-H); IR (KBr,ν/cm-1): 3032, 1668, 1547, 1441, 1311, 1232, 810. Anal. calc for C38H24O8: C 74.99, H 3.97; found C 74.87, H 3.91. MS (FAB):m/e, 608 (M+).
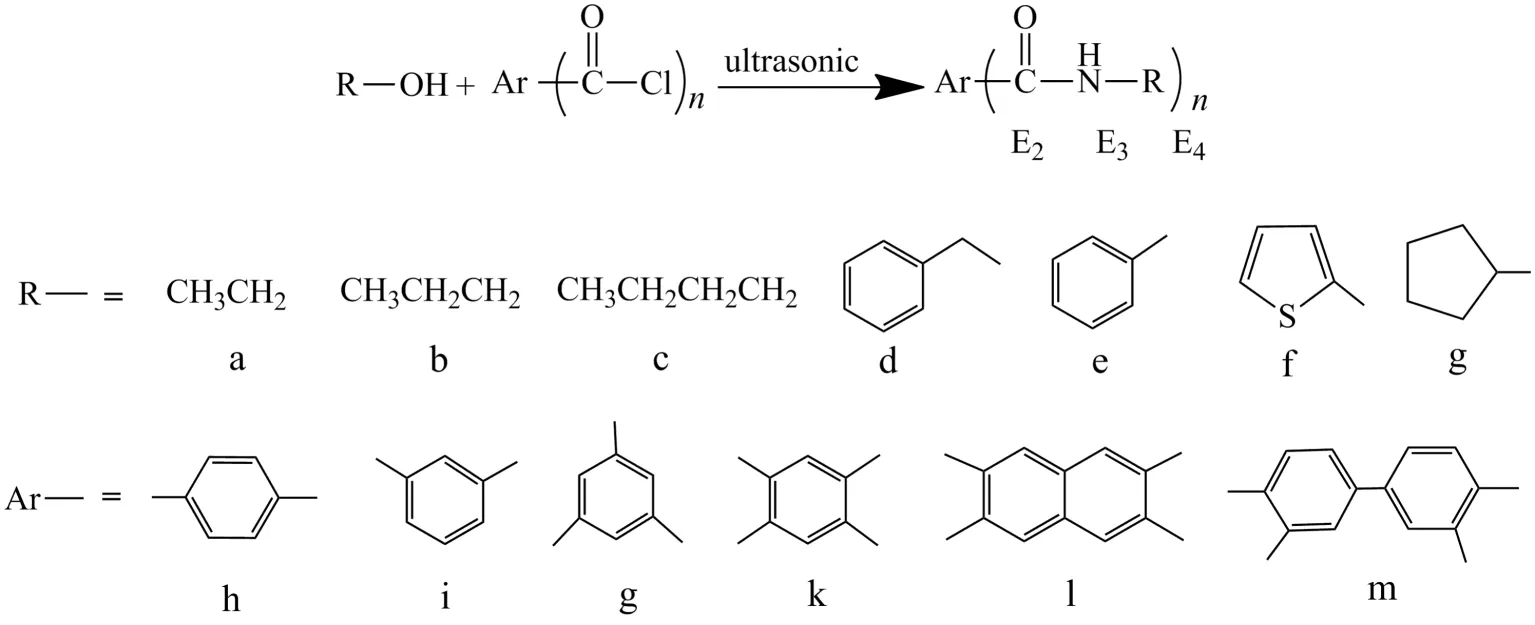
Fig.2 General routes for synthesis of bridged (bis-, tri- and tetra-) aryl esters图2 桥联型(双、三、四)芳基酯类化合物通用合成路线
ProductE4bm: white solid, 79% yield.m.p.262-263 ℃.1H NMR (CDCl3, 400 MHz)δ: 8.86 (s, 2H, Ar-H), 8.47 (s, 2H, Ar-H), 4.44 (m, 6H, C-H), 4.12 (q, 2H, C-H), 2.19 (m, 4H, C-H), 1.67 (m, 5H, C-H), 1.13 (m, 6H, C-H), 0.96 (m, 5H, C-H); IR (KBr,ν/cm-1): 3031, 1658, 1545, 1436, 1305, 1246, 808. Anal. calc for C28H34O8: C 67.45, H 6.87; found C 67.49, H 6.81. MS (FAB):m/e, 498 (M+).
2.4 Amides with the number of groups n=2
The synthetic route of acylation reaction is shown in Fig.3.
ProductA2hp: white solid, 96% yield.m.p.265-266 ℃.1H NMR(CDCl3, DMSO, 400 MHz)δ: 10.43(s, 2H, N-H), 8.10(s, 4H, Ar-H), 7.81(d, 4H, Ar-H), 7.38(t, 4H, Ar-H), 7.13(t, 2H, C-H); IR (KBr,ν/cm-1): 3329, 1648, 1525, 1440, 878. Anal. calc for C20H16N2O2: C 75.93, H 5.10, N 8.86; found C 75.96, H 5.11, N 8.89. MS (FAB):m/e, 316 (M+).
ProductA2ip: white solid, 91% yield.m.p.288-289 ℃.1H NMR(CDCl3, DMSO, 400 MHz)δ: 9.20(t, 2H, N-H), 7.98(s, 4H, Ar-H), 7.33(d, 8H, Ar-H),7.25(m, 2H, Ar-H), 4.50(d, 4H, C-H);IR (KBr,ν/cm-1): 3279, 1631, 1541, 1462, 866. Anal. calc for C22H20N2O2: C 76.72, H 5.85, N 8.13; found C 76.65, H 5.82, N 8.15. MS (FAB):m/e, 344 (M+).
ProductA2jp: white solid, 94% yield.m.p.292-293 ℃.1H NMR(CDCl3, DMSO, 400 MHz)δ:10.50(s, 2H, N-H),8.11(s, 4H, Ar-H), 7.83(d, 4H, Ar-H), 7.22(t, 4H, Ar-H); IR (KBr,ν/cm-1): 3334, 1647, 1522, 1404, 1231, 833. Anal. calc for C20H14F2N2O2: C 68.18, H 4.01, N 7.95; found C 68.17, H 4.02, N 7.97. MS (FAB):m/e, 352 (M+).
ProductA2kp: white solid, 92% yield.m.p.313-314 ℃.1H NMR(CDCl3, DMSO, 400 MHz)δ:10.32(s, 2H, N-H), 8.07(s, 4H, Ar-H), 7.67(d, 4H, Ar-H), 7.17(d, 4H, Ar-H), 2.37(s, 6H, C-H); IR (KBr,ν/cm-1): 3312, 1644, 1518, 1402, 811. Anal. calc for C22H20N2O2: C 76.72, H 5.85, N 8.13; found C 76.78, H 5.82, N 8.15. MS (FAB):m/e, 344 (M+).
ProductA2hq: white solid, 99% yield.m.p.265-266 ℃.1H NMR(CDCl3, DMSO, 400 MHz)δ:10.57(s, 2H, N-H),8.59(s, 1H, Ar-H),8.15(s, 2H, Ar-H), 7.83-7.69(d, 5H, Ar-H), 7.37 (s, 4H, Ar-H),7.13(s, 2H, Ar-H); IR (KBr,ν/cm-1): 3418, 1638, 1546, 1500, 700. Anal. calc for C20H16N2O2: C 75.93, H 5.10, N 8.86; found C 75.91, H 5.15, N 8.86. MS (FAB):m/e, 316 (M+).
ProductA2iq: white solid, 96% yield.m.p.288-289 ℃.1H NMR(CDCl3, DMSO, 400 MHz)δ:9.18(s, 2H, N-H),8.40(s, 1H, Ar-H),8.03(d, 2H, Ar-H),7.58(t, 1H, Ar-H),7.33(d, 8H, Ar-H),7.25 (d, 2H, Ar-H), 4.49(d, 4H, C-H); IR (KBr,ν/cm-1): 3306, 1637, 1542, 1439, 699. Anal. calc for C22H20N2O2: C 76.72, H 5.85, N 8.13; found C 76.74, H 5.81, N 8.15. MS (FAB):m/e, 344 (M+).
ProductA2jq: white solid, 94% yield.m.p.292-293 ℃.1H NMR(CDCl3, DMSO, 400 MHz)δ:10.50(s, 2H, N-H),8.52(s, 1H, Ar-H),8.15(d, 2H, Ar-H),7.82(d, 4H, Ar-H); IR (KBr,ν/cm-1): 3270, 1648, 1524, 1403, 1234, 830. Anal. calc for C20H14F2N2O2: C 68.18, H 4.01, N 7.95; found C 68.16, H 4.07, N 7.93. MS (FAB):m/e, 352 (M+).
ProductA2kq: white solid, 93% yield.m.p.303-304 ℃.1H NMR(CDCl3, DMSO, 400 MHz)δ: 10.35(s, 2H, Ar-H), 8.51(s, 1H, Ar-H), 8.12(d, 2H, Ar-H), 7.69(d, 5H, Ar-H), 7.18(d, 4H, Ar-H), 2.29(s, 6H, C-H); IR (KBr,ν/cm-1):3324, 1648, 1534, 1452, 824. Anal. calc for C22H20N2O2: C 76.72, H 5.85, N 8.13; found C 76.74, H 5.89, N 8.15.MS (FAB):m/e, 344 (M+).
2.5 Amides with the number of groups n=3
The synthetic route of acylation reaction is shown in Fig.3.
ProductA3lr: white solid, 86% yield.m.p.166.5-166.3 ℃.1H NMR(CDCl3, DMSO, 400 MHz)δ:10.17(s, 2H, N-H), 8.29 (s, 1H, Ar-H), 8.18(d, 4H, Ar-H),7.69(d, 2H, Ar-H), 7.12(s, 1H, Ar-H), 7.11(t, 3H, Ar-H); IR (KBr,ν/cm-1): 3202, 3034, 1699, 1525, 1443,823. Anal. calc for C21H33N3O3: C 67.17, H 8.86, N 11.19; found C 67.14, H 8.89, N 11.18. MS (FAB):m/e, 375 (M+).
ProductA3hr: white solid, 83% yield.m.p.244.5-245.3 ℃.1H NMR(CDCl3, DMSO, 400 MHz)δ:10.09 (d, 3H, N-H), 8.29 (d, 3H, Ar-H), 8.11 (t, 6H, C-H), 8.02 (t, 4H, C-H), 7.08(s, 2H, C-H), 6.90 (d,3H, C-H); IR (KBr,ν/cm-1): 3143, 3042, 1744, 1565,1483,801. Anal. calc for C27H21N3O3: C 74.47, H 4.86, N 9.65; found C 74.44, H 4.87, N 9.62. MS (FAB):m/e, 435 (M+).
ProductA3mr: white solid, 91% yield. m.p.224.5-225.3 ℃.1H NMR(CDCl3, DMSO, 400 MHz)δ: 10.09 (d, 3H, N-H), 8.29 (d, 3H, Ar-H), 8.11 (t, 6H, C-H), 8.02 (t, 4H, C-H), 7.08 (s, 2H, C-H),6.90 (d,4H, C-H); IR (KBr,ν/cm-1): 3153, 3022, 1714, 1565, 1483, 816. Anal. calc for C27H21N3O3S2: C 55.61, H 3.33, N 9.26; found C 55.65, H 3.31, N 9.25. MS (FAB):m/e, 453 (M+).
2.6 Amides with the number of groups n=4
The synthetic route of acylation reaction is shown in Fig.3.
ProductA4ns: light yellow solid, 76% yield.m.p.254.6-255.2 ℃.1H NMR(CDCl3, DMSO, 400 MHz)δ: 10.09 (d, 4H, N-H), 8.29 (d, 3H, Ar-H),7.29 (s, 3H, C-H); IR (KBr,ν/cm-1): 3133, 3032, 1744, 1568, 1491, 810 . Anal. calc for C30H42N4O4: C 68.94, H 8.10, N 10.72; found C 68.94, H 8.14, N 10.7. MS (FAB):m/e, 522 (M+).
ProductA4ht: yellow solid, 96% yield.m.p.264.1-265.9 ℃.1H NMR(CDCl3, DMSO, 400 MHz)δ: 10.09 (d, 4H, N-H), 8.29 (d, 4H, Ar-H), 8.29 (d,4H,Ar-H), 8.29 (s, 4H, Ar-H), 8.29 (t, 4H, Ar-H); IR (KBr,ν/cm-1): 3123,3022,1754,1578,1511,815.Anal. calc for C38H28N4O4: C 75.48,H 4.67, N 9.27; found C 75.45, H 4.63, N 9.29. MS (FAB):m/e, 604 (M+).
ProductA4ou: grayish white solid, 81% yield.m.p.264.1-265.9 ℃.1H NMR(CDCl3, DMSO, 400 MHz)δ: 10.09 (d, 4H, N-H), 8.29 (t, 6H, Ar-H), 4.33 (s, 2H, C-H), 4.29 (s, 4H, C-H), 4.11(s, 2H, C-H); IR (KBr,ν/cm-1): 3143, 3112, 1746, 1534, 1498, 803. Anal. calc for C28H38N4O4: C 67.99, H 7.77, N 11.33; found C 67.96, H 7.78, N 11.34. MS (FAB):m/e, 494 (M+).

Fig.3 General route for synthesis of bridged (bis-, tri- and tetra-) aryl amides图3 桥联型(双、三、四)芳基酰胺类化合物通用合成路线
3 Analysis and discussion
3.1 Physical properties and elemental analysis
Elemental analysis (data shown in Tab.1):The C and H of the target compound were consistent with the theoretical composition.
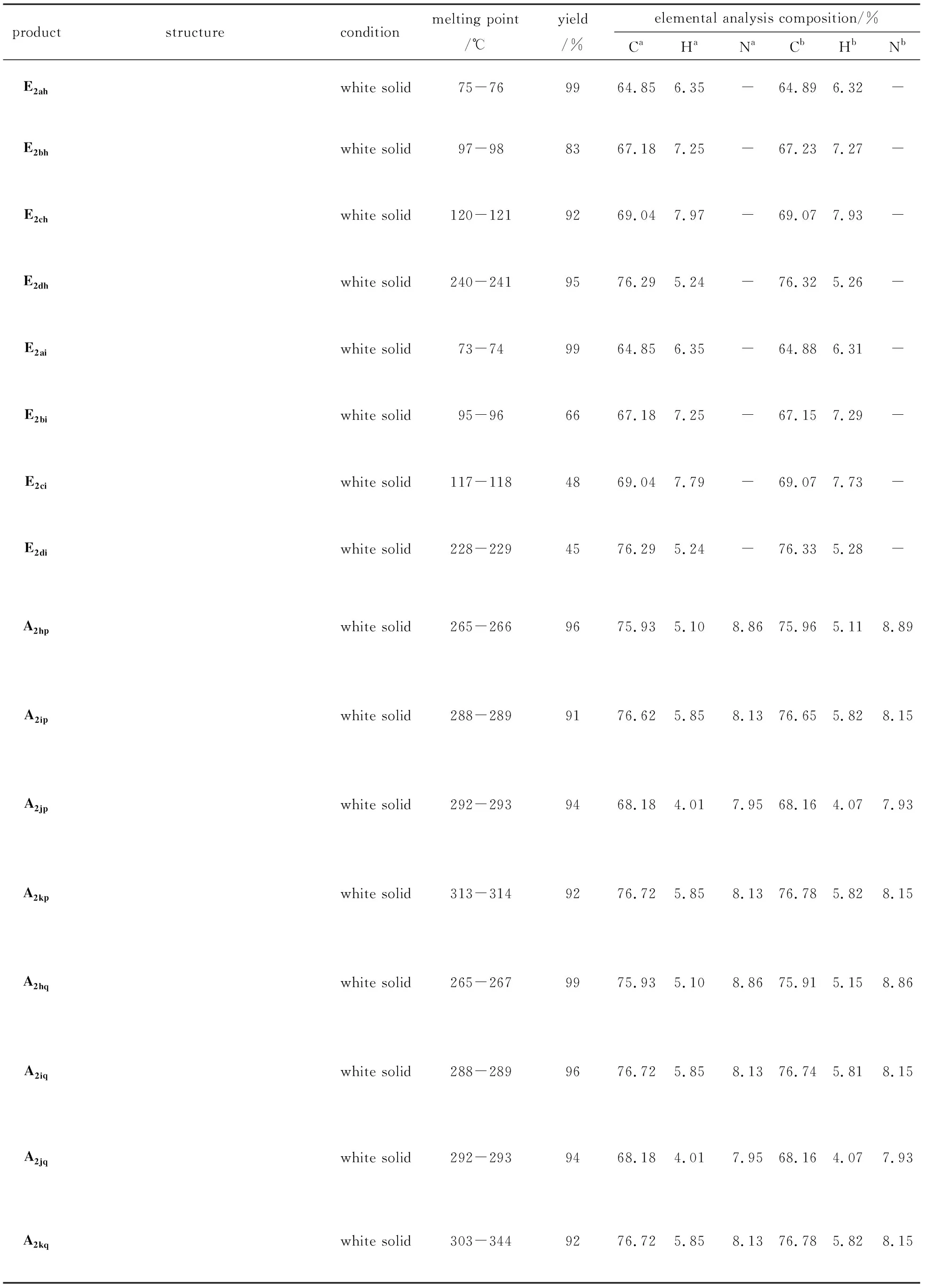
Tab.1 Physical property constants and elemental analysis compositions of the products表1 产物的物性常数及元素分析组成
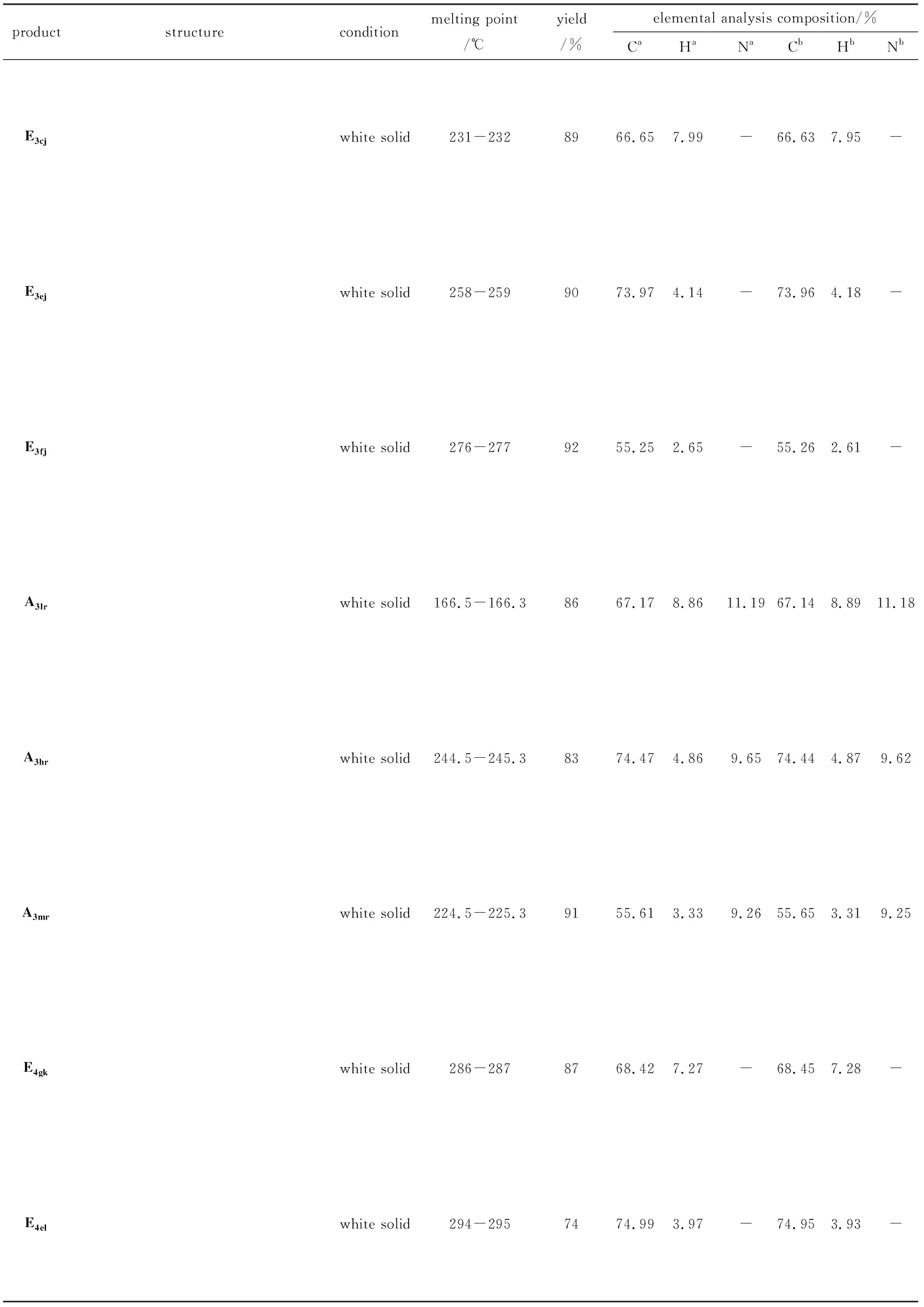
productstructureconditionmelting point/℃yield/%elemental analysis composition/%CaHaNaCbHbNbE3cjwhite solid231-2328966.657.99-66.637.95-E3ejwhite solid258-2599073.974.14-73.964.18-E3fjwhite solid276-2779255.252.65-55.262.61-A3lrwhite solid166.5-166.38667.178.8611.1967.148.8911.18A3hrwhite solid244.5-245.38374.474.869.6574.444.879.62A3mrwhite solid224.5-225.39155.613.339.2655.653.319.25E4gkwhite solid286-2878768.427.27-68.457.28-E4elwhite solid294-2957474.993.97-74.953.93-

productstructureconditionmelting point/℃yield/%elemental analysis composition/%CaHaNaCbHbNbE4bmwhite solid262-2637967.456.87-67.486.86-A4nalight yellow solid254.6-255.27668.948.1010.7268.948.1410.70A4htyellow solid264.1-265.99675.484.679.2775.454.639.29A4ougrayish white solid264.1-265.98167.997.7411.3367.967.7811.34
3.2 Infrared and mass spectrometry analysis
In the IR spectra of the target compound(data shown in Tab.2), each characteristic absorption peak of the compounds was confirmed. For instance, the stretching vibration of the amine N-H was about 3300-3100 cm-1; the stretching vibration of the aromatic C-H was about 3100-2800 cm-1; the stretching vibration of the ester -CO was about 1700-1600 cm-1; the stretching vibration of benzene ring skeleton was about 1690-1400 cm-1; the stretching vibration of the ester C-O-C was about 1400-1100 cm-1; the Out-of-plane bending vibration of the aromatic ring was about 850-700 cm-1. According to the characteristic vibration peaks of N-H, C-O and benzene rings, the structure of the product was consistent with that of the speculated structure. In MS spectra, the molecular fragment peaks of the compounds were consistent with the molecular theory.

Tab.2 IR and MS date of the product表2 产物红外和质谱数据
3.3 1H NMR analysis
In the1H NMR spectra (data shown in Tab.3), the chemical shift of N-H on the amine was about 10-9, such asA2hz10.43(s, 2H, N-H),A3lr10.17(s, 2H, N-H)andA4ns10.09 (d, 4H, N-H).The chemical shift of hydrogen on benzene ring was about 8-7,E2ah8.10(s, 4H, Ar-H). The chemical shift of methyl hydrogen was 1.5-1, for example, the chemical shift ofE2ahmethyl hydrogen was 1.42 (t, 6H, C-H); and 5.31 (s, 4H, C-H), 5.32 (s, 4H, C-H) were assigned to the methylene hydrogen inE2dhandE2di ,respectively.The chemical shifts of hydrogen in benzylmethylene of amidesA2ipandA2iqbenzylidene were observed at 4.50 (d, 4H, C-H), 4.49 (d, 4H, C-H).

Tab.3 1H NMR data of product表3 产物1H NMR数据
3.4 Conjecture of reaction mechanism(n=2)
Under ultrasonic irradiation, ultrasonic wave directly interacted with reaction molecules. Due to the effect of ultrasonic cavitation and secondary effect, the reaction system generated local high temperature and high pressure, and at the same time produced strong shock wave, which promoted the collision and aggregation between molecules, thus speeding up the reaction process. The possible reaction mechanism of etherification and acylation was shown in Fig.4 and Fig.5. In the process, the alcohol (amine) acted as nucleophilic reagent. Hydroxy oxygen and amino nitrogen attacked carbonyl carbon to form negative oxygen ions, then the intermediate I and III were separated from one molecule of HR3 to form intermediates II and IV, respectively. Nucleophilic addition of another molecule alcohol or amine followed subsequently, and the process was repeated to obtain esters or amide derivatives.
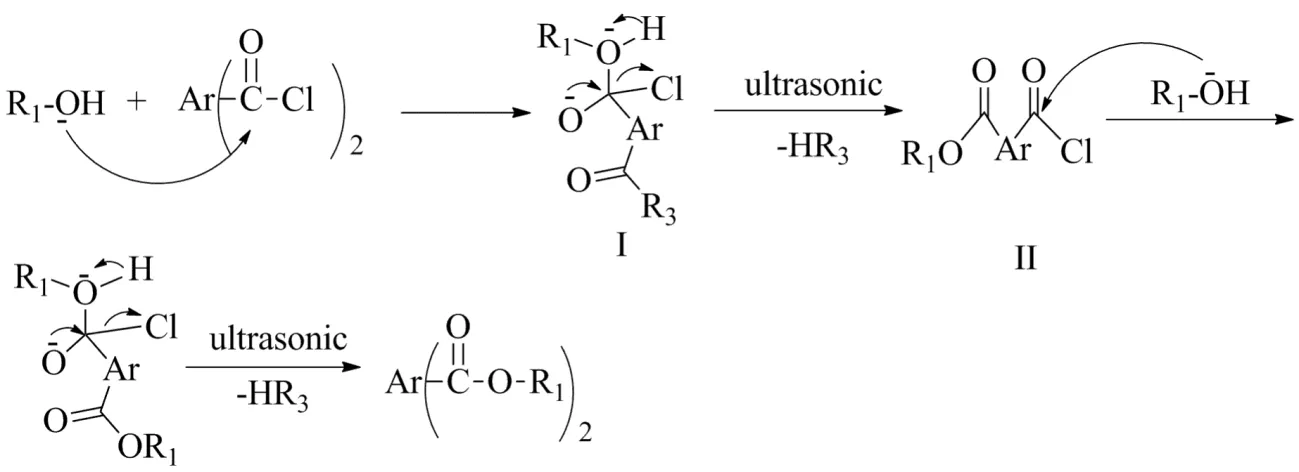
Fig.4 Esterification reaction mechanism of bridged (bis-, tri- and tetra-) aryl amides图4 桥联型(双、三、四)芳基酯类化合物的酯化反应机理

Fig.5 Acylation reaction mechanism of bridged (bis-, tri- and tetra-) aryl amides图5 桥联型(双、三、四)芳基酯类化合物的酰化反应机理
3.5 Optimization of reaction experiment conditions
3.5.1 Determination of the optimum ratio of raw materials(n=2)
The reaction of ethanol and aniline withp-benzoyl chloride for the synthesis of compoundsE2ah/A2hpwere studied as a model. The reaction was conducted at room temperature under ultrasonication for 20/15 min, the amount of terephthaloyl chloride was fixed at 0.25 mmol, while the ratio of ethanol/aniline to chloride were varied from 2∶1 to 2.2∶2. The increase of the ratio was beneficial to the equilibrium shift of esterification and acylation reaction to the right; however, as can be seen from Tab.4, a maximum yield was obtained at a ratio of 2.05∶1, exceeding which the yield decreased. This was not totally surprising, since the further increase of ratio resulted in more deviation from stoichiometry, the yield would decrease.

Tab.4 Effect of raw material ratio on esterification and acylation reaction表4 原料配比对酯化和酰化反应的影响
3.5.2 Ultrasonic radiation time
To determinate the optimum ultrasonic time of ester/acylation reaction, with synthesis of compoundE2ahwas studied in detail. The amount ofp-benzoyl chloride and ethanol/aniline was 0.25 mmol and 0.51 mmol respectively. From Tab.5, it could be seen that the increase of reaction time could improve the yield, but too long reaction time resulted in the decrease of yield. The esterification and acylation reaction was a reversible reaction. Before the reaction was complete, the increase of reaction time increased the yield. However, after the reaction was completed, continued ultrosonication might lead to side reactions and decrease the yield. The optimum reaction time was 20 minutes for esterification and 15 minutes for acylation .

Tab.5 Effect of ultrasonic time on esterification and acylation reaction表5 超声时间对酯化和酰化反应的影响
3.6 Advantages of this method
There are many methods for the synthesis of monoamides and monoesters, which generally involve heating/refluxing, using alkali (including organic and inorganic base) or acid (including organic and inorganic acid) as catalyst. However, production ofbis-,tri- andtetra- amides involves the formation of multiple ester bonds, which is much more complicated. In the present work, two kinds of carboxylic acid derivatives,bis-,tri-,tetra- ester and amide, were synthesized by ultrasonic radiation, with no need for prolonged heating and refluxing, or use of a catalyst. It took only 20 min or 15 min to complete the reaction.
Moreover, compared with the traditional synthesis method, the ultrasonic radiation method made use of the “cavitation effect” and “secondary effect” of ultrasonic radiation, which produced instantaneous high temperature and high pressure to initiate chemical reaction, formed local high energy center, allowed the reaction solution to stir violently, and greatly shortened the reaction time.The product was also directly precipitated from the solvent, thereby avoided complicated separation process. In addition, no catalyst was used, especially the highly polluting acid or alkaline reagent. The solvent used was non-toxic dichloromethane. The method simplified the synthesis route and saved cost, and was a good general method for synthesizing aryl carboxylic acid derivatives.
4 Conclusion
28 bridged aryl carboxylic acid derivatives were synthesized at room temperature by the reaction of alcohol and amine with acyl chloride using ultrasonic radiation. The structure of the compounds was confirmed by IR,1H NMR, MS and elemental analysis. The optimum reaction conditions were obtained. Compared with the traditional methods, this approach not only can be used for the efficient synthesis of thebis-,tri- andtetra-arylcarboxylic acid derivatives in one step, but also has many advantages such as ease of operation, less pollution, simple post-treatment, low cost, etc. It is a new, efficient general method for the synthesis of bridged arylcarboxylic acid derivatives and has great potential for reducing the cost of some industrially important fine chemicals.
