超高效合相色谱-串联质谱法测定食用植物油和油条中15种3-氯-1,2-丙二醇脂肪酸酯
2020-11-02杨光勇郭苍亭郭金喜
杨光勇, 郭苍亭, 薛 光, 郭金喜
(1. 新疆维吾尔自治区产品质量监督检验研究院, 新疆 乌鲁木齐 830011;2. 乌鲁木齐市疾病预防控制中心, 新疆 乌鲁木齐 830026)
3-氯-1,2-丙二醇脂肪酸酯(3-chloro-1,2-propanediol fatty acid esters, 3-MCPDE)的危害和污染状况近年受到国际社会的广泛关注。3-MCPDE主要存在于食用植物油中[1,2],且大多产生于精炼过程,尤其是脱臭步骤[3,4],形成机制尚不明确。此外,食品在烹饪、加工过程中也会生成3-MCPDE[5,6],尤其是加入氯化物后,3-MCPDE的含量会急剧上升[7,8]。随着研究范围的不断扩大,研究者发现3-MCPDE广泛存在于奶粉[9]、油炸食品[10]、牛奶[11]、谷物[12]等多种食品及其原材料中。我国于2015年开始将3-MCPDE纳入食品安全风险监测计划,近几年相关研究[13-16]及监测数据显示,国内3-MCPDE的整体检出率居高不下,污染状况不容乐观。
3-MCPDE的毒性目前仍不清楚,但其代谢产物3-氯-1,2-丙二醇(3-MCPD)被证明具有生殖毒性、肾脏毒性、潜在的致癌性及免疫抑制性[17,18],是氯丙醇类化合物中危害性最大的物质,常被作为氯丙醇的代表和毒性参照物[1]。3-MCPD具有两个羟基和两种光学异构体,可与多种脂肪酸反应形成各类3-MCPD单酯和二酯,结构上的差异使其在人体内的酶解速率、代谢途径、毒理学作用等方面表现出极大差异[19,20]。因此,有必要对各3-MCPDE单体进行分离和测定,为评估3-MCPDE的健康风险提供更加准确的数据。
目前,3-MCPDE的检测方法有间接法和直接法。间接法是将样品水解后,提取出水解液中的3-MCPD进行衍生,用气相色谱-质谱法分析其衍生物[21,22]。间接法只能测定氯丙醇酯总量,无法确定其单体的种类和含量。方法操作烦冗复杂,所用衍生试剂昂贵且不稳定;水解过程还会将油脂中的缩水甘油酯转化成氯丙醇,影响测定结果的准确性[23]。直接法是在不破坏3-MCPDE结构的情况下,采用液相色谱法[24]、液相色谱-质谱法[25,26]、超临界流体色谱-质谱法[27]等测定其单体含量的方法,其中有些采用溶解后直接进样分析的方法[27,28],但大部分是先净化再检测[29-31]。液相色谱法定性能力不足,灵敏度低,无法测定痕量3-MCPDE。基于反相液相色谱的质谱方法,流动相与样品不能很好兼容,色谱柱易堵塞,柱效下降极快;未经净化或净化不充分时质谱仪极易污染,基质干扰明显,尽管使用了氘代化合物作为内标,但某些研究的回收率仍很低(≥62.55%)[27,32];一些研究[28,32]使用含钠盐的流动相,严重污染和腐蚀离子源,需每天清洗甚至更换组件,大幅增加检测成本。
超高效合相色谱(ultra performance convergence chromatography, UPC2)兼具超临界流体色谱的优势和传统高效液相色谱的易用性,已被成功应用于甘油三酯(TAGs)[33]、游离脂肪酸(FFAs)[34]、塑化剂[35]等脂溶性化合物的分析。本实验用超高效合相色谱-三重四极杆质谱建立了测定食用植物油和油条中15种3-MCPDE单体含量的方法,为测定植物油及油炸食品中氯丙醇酯类化合物提供了绿色环保、快速准确的新方法。该法灵敏度高,专属性好,基质兼容性强,较传统方法极具优势,能有效测定3-MCPDE的组成和含量,对其毒理学评价、形成机制研究等具有重要意义。
1 实验部分
1.1 仪器与试剂
UPC2-TQS超高效合相色谱-三重四极杆质谱仪、ACQUITY QDa质谱检测器(美国Waters公司), N-EVAP-24氮吹仪(美国Organomation公司),涡旋混合器(德国Heidolph公司);氨基吸附剂(40 μm,美国Agilent公司)。
标准品:1-棕榈酸-3-氯-1,2-丙二醇酯(P)、1-硬脂酸-3-氯-1,2-丙二醇酯(St)、1-油酸-3-氯-1,2-丙二醇酯(O)、1-亚油酸-3-氯-1,2-丙二醇酯(L)、1-亚麻酸-3-氯-1,2-丙二醇酯(Ln)、1,2-二棕榈酸-3-氯-1,2-丙二醇酯(PP)、1,2-二硬脂酸-3-氯-1,2-丙二醇酯(StSt)、1,2-二油酸-3-氯-1,2-丙二醇酯(OO)、1,2-二亚油酸-3-氯-1,2-丙二醇酯(LL)、1,2-二亚麻酸-3-氯-1,2-丙二醇酯(LnLn)购自日本和光公司;1-月桂酸-3-氯-1,2-丙二醇酯(La)、1-肉豆蔻酸-3-氯-1,2-丙二醇酯(My)、1-油酸-2-亚油酸-3-氯-1,2-丙二醇酯(OL)、1-油酸-2-亚麻酸-3-氯-1,2-丙二醇酯(OLn)、1-油酸-2-硬脂酸-3-氯-1,2-丙二醇酯(OSt)购自加拿大TRC公司;甲酸、正己烷、甲醇、乙腈、乙酸乙酯、异丙醇均为色谱纯,购自美国Thermo Fisher公司。食用植物油和油条样品均购自当地市场。
1.2 实验方法
1.2.1仪器条件
色谱条件:色谱柱为Viridis HSS C18 SB柱(150 mm×2.1 mm, 1.8 μm);柱温为45 ℃;流动相A为超临界CO2, B为40%乙腈甲醇溶液(含0.1%甲酸);流速为1.0 mL/min。洗脱梯度程序为0~1.0 min, 0.2%B; 1.0~9.0 min, 0.2%B~2.0%B; 9.0~11.0 min, 2.0%B~25%B; 11.0~13.0 min, 25%B; 13.0~14.0 min, 25%B~0.2%B。系统背压为12.07 MPa;补偿液为97%异丙醇水溶液(含0.2%氨水);流速为0.2 mL/min;进样体积为5 μL。
质谱参数:电喷雾电离源,正离子模式(ESI+);采集方式为多反应监测(MRM)模式;离子源温度150 ℃;去溶剂气流量750 L/h;去溶剂气温度350 ℃;毛细管电压3.5 kV;流动相切换时间设置为4~9 min切换至ESI源,其他时间段排至废液。15种3-MCPDE的保留时间、相对分子质量、母离子和子离子见表1。
ACQUITY QDa质谱检测器操作简单直观,抗污染能力强,实验将其与超高效合相色谱仪联用后用于优化样品制备条件。
ACQUITY QDa质谱检测器参数为:正离子模式,扫描范围300~910 Da,离子源温度150 ℃,去溶剂气温度300 ℃,毛细管电压1.5 kV,锥孔电压15 kV,增益1,采集速率1点/s;负离子模式,扫描范围180~400 Da,毛细管电压0.8 kV,采集速率2点/s,其他参数同前。
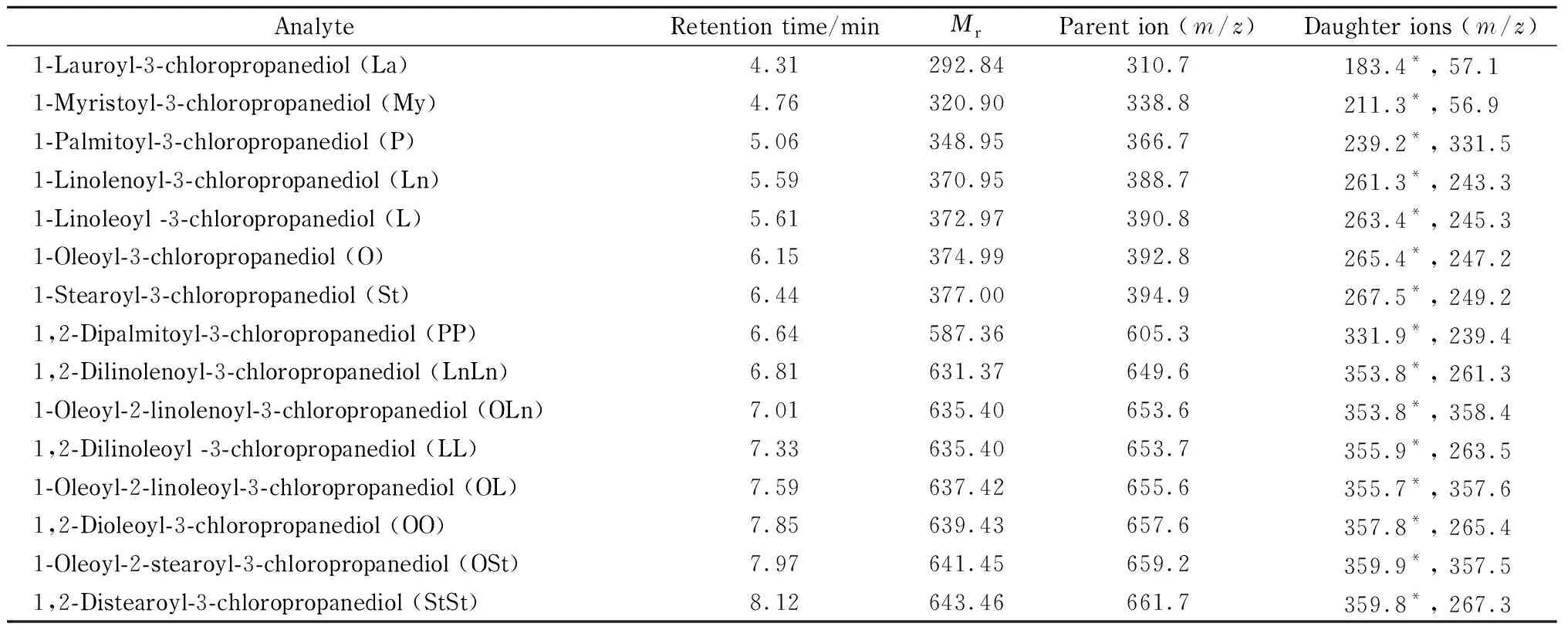
表 1 15种3-MCPDE的保留时间、相对分子质量、母离子及子离子
1.2.2样品溶液的制备
标准品溶液:精密称取适量各标准品,用正己烷制成1 g/L的储备液;再量取各储备液适量,用正己烷稀释成1 mg/L的标准使用液,上述溶液均置于-18 ℃冰箱保存。
样品溶液:将植物油充分混匀备用,油条按GB 5009.6-2016《食品中脂肪的测定索氏提取法》提取出油脂后备用;称取2.00 g氨基吸附剂,以正己烷为溶剂湿法装入自制层析柱(经改造的10 mL酸式滴定管),临用前排出正己烷。
称取0.100 g样品,置于1.5 mL离心管中,加入0.2 mL正己烷,涡旋溶解后转入层析柱,以0.4 mL正己烷分两次洗涤离心管,洗涤液一并转入层析柱;用20 mL正己烷洗脱,控制流速为3 mL/min,收集第7~14 mL洗脱液,其余弃去;再以10 mL正己烷-乙酸乙酯(6∶4, v/v)洗脱,流速同上,收集第3~9 mL洗脱液。合并收集的洗脱液,置于离心管中,于60 ℃氮吹至干,加入1 mL正己烷-异丙醇(98∶2, v/v),涡旋溶解残渣后,过0.22 μm滤膜。
2 结果与讨论
2.1 色谱和质谱条件的优化
2.1.1色谱柱的选择
因未酯化羟基和碳链数的影响,使3-MCPD单酯和双酯的分离较为容易,但各单酯、双酯单体之间的结构和极性极为相似,为选择色谱柱带来一定难度。实验考察了3种填料的UPC2色谱柱Viridis HSS C18 SB(150 mm×2.1 mm, 1.8 μm)、Torus 1-AA(100 mm×3.0 mm, 1.8 μm)、BEH(100 mm×3.0 mm, 1.8 μm)对分离效果的影响。HSS C18 SB色谱柱是经过改进的反相色谱柱,对酯类化合物具有很好的保留能力;1-AA色谱柱填料在硅胶颗粒上键合了1-氨基蒽,可与3-MCPDE的不饱和酯链产生电子转移、π-π等多种作用,具有较好的选择性;BEH色谱柱填装了亚乙基桥杂化颗粒,对于各3-MCPDE单体间的极性差异较为敏感。然而选用1-AA色谱柱无法有效分离15种3-MCPDE,各组分色谱峰均互相包埋或形成肩峰;选用BEH色谱柱虽能分离3-MCPD单酯和双酯,但对于极性差异较小的目标化合物仍无法实现基线分离;选用HSS C18 SB色谱柱时,各3-MCPDE分离趋势最为明显。因此,实验选择HSS C18 SB色谱柱进行测定。
2.1.2色谱柱温度及系统背压的选择
色谱柱温度和系统背压的变化会改变超临界CO2的密度,从而影响其洗脱能力,当柱温升高、背压减小时,流动相密度变小,化合物保留时间延长。实验分别考察了不同柱温(25、35、45、55 ℃)、不同背压(12.07、12.76、13.45、14.13 MPa)对分离效果的影响。结果表明,背压对分离的影响较小;柱温较高时,分析时间延长,柱温较低时,OSt和StSt的色谱峰略有拖尾。综合考虑分离度、系统压力极限等因素,选取45 ℃柱温、12.07 MPa背压为测定条件。
2.1.3流动相改性剂的选择
为改变超临界CO2对化合物的溶解和洗脱能力,需在流动相中引入适量甲醇、乙腈等溶剂作为改性剂。实验分别以不同体积分数(0、10%、20%、30%、40%)的乙腈甲醇溶液作为改性剂,考察了改性剂洗脱强度对分离效果的影响;同时,为增强未键合硅醇羟基和氯丙醇酯的氢键作用,增加色谱选择性,向各改性剂中加入0.1%(v/v)的甲酸。结果表明,随着乙腈体积分数的增加,洗脱强度变弱,各组分保留时间延长,分离趋势增加;当乙腈体积分数为40%时,各组分基本达到基线分离(分离度≥1.33)。
在优化的色谱条件下,采用UPC2-MS/MS对15种3-MCPDE混合标准溶液(1 mg/L)进行测定,其提取离子流色谱图见图1。
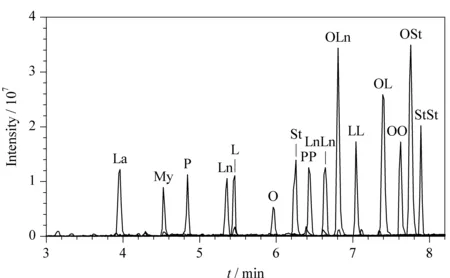
图 1 15种3-MCPDE混合标准溶液(1 mg/L)的提取离子流色谱图Fig. 1 Extracted ion current chromatogram of the 15 3-MCPDE mixed standard solution (1 mg/L)

图 2 补偿液中(a)氨水和(b)纯水的体积分数对4种3-MCPDE峰高的影响Fig. 2 Effects of volume fractions of (a) ammonium hydroxide and (b) water in compensation solution on the peak heights of the four 3-MCPDE
2.1.4补偿液组成及流速的优化
补偿液由柱塞泵输送至系统中,主要用于传输化合物至质谱仪,并促使其离子化。补偿液不参与色谱分离,但其流速和组成对化合物色谱峰形、离子化效率等均有较大影响。为获得较好的色谱峰形和响应强度,实验选用溶解性能和促离子化能力较为折中的异丙醇作为补偿液,并在异丙醇中加入添加剂以促进3-MCPDE离子加合物的形成。尚月[36]的研究证实,3-MCPDE的铵加合物比其他加合物形式具有更好的质谱响应,因此,实验以P、O、PP、OO为对象考察了含不同体积分数(0.1%、0.2%、0.3%、0.4%)氨水的异丙醇补偿液在不同流速(0.1、0.2、0.3、0.4 mL/min)下对测定结果的影响。结果表明,当补偿液流速为0.2 mL/min、氨水体积分数为0.2%(v/v)时目标化合物色谱峰形和响应可达到最佳状态(见图2a)。此外,实验发现在含0.2%(v/v)氨水的异丙醇补偿液中加入适量的纯水可进一步提高方法的灵敏度,通过比较添加不同体积分数(1%、2%、3%、4%、5%、6%)纯水时化合物的峰高(见图2b),可知纯水的最佳体积分数为3%;当超过5%后,系统压力波动明显,设备无法就绪,可能因为水的高比热容使系统动态背压阀无法正常控温,或管路中形成双相流动相。
2.2 样品净化条件的优化
实验曾将加标植物油样品(1 mg/kg)溶解后直接进样分析,但个别组分基质效应(ME)明显,回收率较低(≥34.8%)。通过分析共流出组分的一级、二级质谱图得知,干扰组分主要为FFAs和短链TAGs。因此,实验在傅武胜等[37]研究的基础上,进一步优化了样品前处理条件,制作洗脱曲线以确定收集体积,减小基质干扰。
按1.2.2节流程制备样品,每1 mL收集一次洗脱液,采用1.2.1节参数进行扫描,并对3-MCPDE、TAGs及FFAs的提取离子流色谱图进行积分,绘制体积段和各组分峰面积的洗脱曲线。TAGs和FFAs母离子的m/z分别参考其他文献[34,38]。结果表明,尽管3-MCPDE与TAGs、FFAs存在部分共洗脱现象,但该法仍可去除大部分杂质,有效减小了基质干扰和设备污染,同时提高了方法灵敏度。根据洗脱曲线确定的收集体积为:3-MCPD二酯7~14 mL, 3-MCPD单酯3~9 mL。
2.3.1基质效应
分别配制系列基质匹配标准溶液和系列空白溶剂标准溶液,然后进行测定,按如下公式考察基质效应:ME=|(基质匹配标准曲线斜率/空白溶剂标准曲线斜率-1)|×100%, ME值≤20%为弱基质效应,ME值为20%~50%为中等基质效应,ME值>50%为强基质效应。结果表明,植物油和油条样品的ME值为2.1%~14.6%,基质效应较弱,可以使用溶剂标准曲线进行定量。
2.3.2线性范围、检出限和定量限
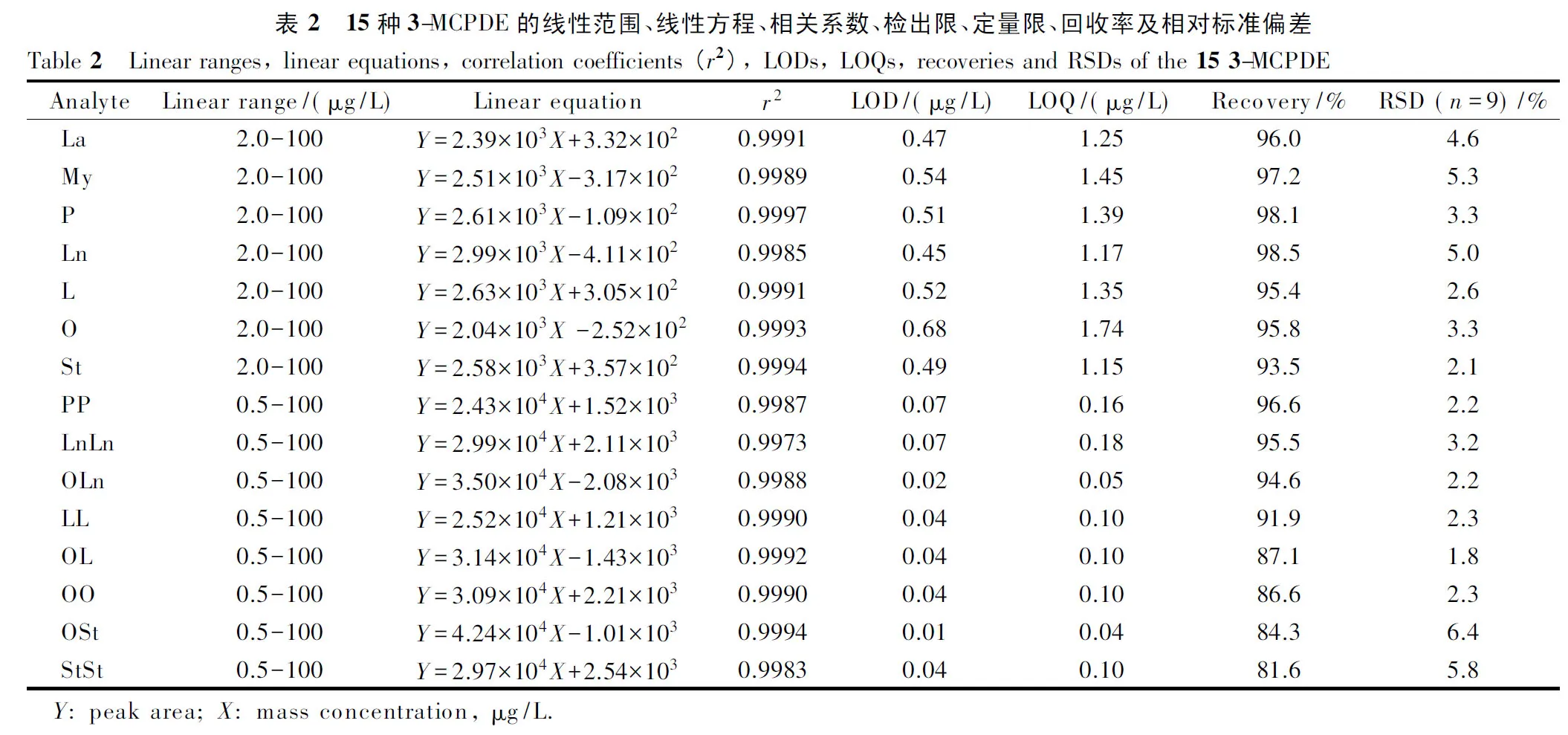
用正己烷-异丙醇(98∶2, v/v)配制各组分质量浓度均为0.5、2.0、5.0、10、25、50、100 μg/L的系列混合标准溶液,经UPC2-MS/MS测定后,以目标物定量离子峰面积为纵坐标(Y)、对应质量浓度为横坐标(X, μg/L)绘制标准曲线。结果表明,15种3-MCPDE在各自的浓度范围内线性关系良好,相关系数(r2)≥0.997 3(见表2)。
在阴性植物油样品添加适量混合标准溶液(1 mg/L),使最终测试溶液中各目标化合物信噪比(S/N)分别为3和10,计算检出限(LOD)和定量限(LOQ)。结果如表2所示,15种3-MCPDE的LOD和LOQ分别为0.01~0.68 μg/L和0.04~1.74 μg/L,低于其他方法[22,24,28]。
2.3.3回收率、精密度
选取阴性植物油样品,加入适量标准储备液,制备成各组分含量均为100、250和1 000 μg/kg的加标样品,每个加标水平制备3份,按1.2.2节前处理方法处理后进行测定,得到样品的平均回收率为81.6%~98.5%(n=9),相对标准偏差为1.8%~6.4%(n=9)(见表2)。
2.4 实际样品的测定
按照建立的方法对44份植物油和8份油条样品进行检测。选取的葵花籽油、棉籽油、红花籽油、菜籽油、花生油等5类44份食用植物油中共有37份检出3-MCPDE,总体检出率为84.1%,检出含量为0.024~4.481 mg/kg;其中菜籽油检出率高达100%,可能与其特殊的加工工艺有关;植物油中检出率最高的3-MCPD单酯、3-MCPD二酯分别为1-硬脂酸-3-氯-1,2-丙二醇酯和1-油酸-2-亚油酸-3-氯-1,2-丙二醇酯,检出含量分别为0.021~0.076 mg/kg和0.008~3.174 mg/kg,这与各文献[25,26,29]报道情况基本一致。
8份油条的总检出率为87.5%,检出含量为0.018~1.144 mg/kg;油条中检出率最高的3-MCPD单酯、3-MCPD二酯分别为1-亚油酸-3-氯-1,2-丙二醇酯和1-油酸-2-亚油酸-3-氯-1,2-丙二醇酯,检出含量分别为0.021~0.053 mg/kg和0.011~0.988 mg/kg,该结果与棉籽油的检出情况较为接近,可能因为烹制样品时选用了价格低廉的棉籽油。
实验还检出一些疑似氯丙醇酯的组分,因无相应标准品而无法准确定量。如保留时间为7.26 min的化合物(见图3a),其母离子m/z为629.1(见图3b),推导其准分子离子为[C37H67O4Cl+NH4]+,含2个C=C键。
二级质谱图如图3c所示,m/z为355.7和331.9的二级碎片离子分别为母离子脱去棕榈酸铵和亚油酸铵后形成的基团,m/z为263.3和239.2的碎片离子分别为亚油酸、棕榈酸C-O单键断裂后形成的亚油酰基团和棕榈酰基团,因此推测该化合物可能为1-棕榈酸-2-亚油酸-3-氯-1,2-丙二醇酯或其位置异构体。
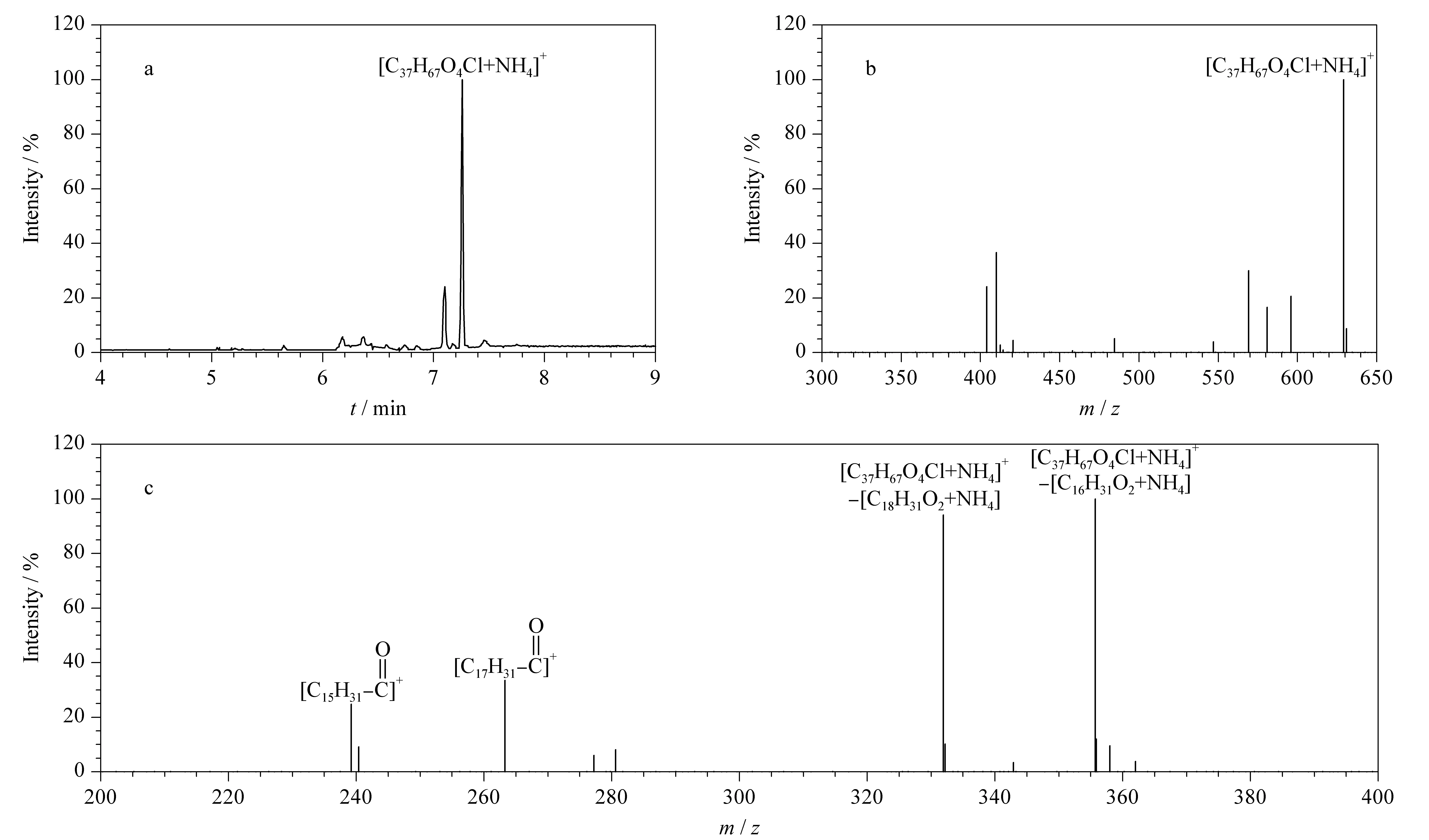
图 3 实际样品中保留时间为7.26 min化合物的(a)提取离子流色谱图、(b)一级质谱图和(c)二级质谱图Fig. 3 (a) Extracted ion current chromatogram, (b) primary mass spectrum and (c) secondary mass spectrum of the compound whose retention time is 7.26 min in the real sample
3 结论
本文建立了UPC2-MS/MS测定植物油和油条中15种3-MCPDE含量的方法,采用优化的样品前处理方式,有效减小了基质干扰,并提高了检测灵敏度。
方法快速准确,灵敏度高,专属性好,绿色环保,所用流动相扩散系数高,质量转移能力强,基质兼容性好,能直接测定植物油和油条中各3-MCPDE单体的种类和含量。同时,该法在积累大量的、更为广泛的分析数据后还具有辨别地沟油的潜力。
但该法仍存在一定不足,主要表现为:所用质谱仪的分辨率有限,对未知3-MCPDE的定性结果可能存在偏差;定量过度依赖价格昂贵且种类有限的单体标准品,而自制标准品难度较大,纯度也无法保证;3-MCPD单酯的离子化效率较双酯低,可能与未酯化羟基有关,后期研究将验证加入酰氯试剂等使其酯化以提高质谱响应的可能性。
