稀土La掺杂对新型二维材料磷烯光电性质的影响
2020-09-17李湘环张忠政张春红
李湘环 张忠政 张春红 张 琦
(1、4.安顺学院数理学院,贵州 安顺561000)(2、3.安顺学院数理学院;安顺学院航空电子电气与信息网络工程中心,贵州 安顺561000)
0 引言
随着科技的快速发展,传统材料已经不能满足高性能制造材料的需求,因此人们在不断寻找新型材料来替代传统材料。二维材料是新材料研究的一个新方向。磷烯作为新型的二维半导体材料,具有优越的光电特性,所以在光电子器、气体传感器和太阳能电池等制造材料方面前景很好。
于是,学者们对磷烯开展了深入的研究。磷烯是由单元素磷构成的,呈蜂窝褶皱状结构的新型二维半导体材料,具有0.59eV~1.51eV可调节的直接带隙,而且有载流子迁移率高、导电导热能力良好、漏电流调制率高、各向异性等性质[1-7]。随后,学者们对磷烯的掺杂改性也做了一些研究,得到的结论有:采用Al,Si,S和Cl掺杂磷烯的几何结构等性质做了研究,发现只有Al掺杂磷烯后仍没有磁性,而Si,S,Cl掺杂后磷烯为金属特性且具有磁性[8]。关于碱金属元素、过渡金属元素和非金属元素原子掺杂磷烯进行电学和磁性调控的研究已有报道[9-17],而有关稀土元素掺杂磷烯光电性质影响的理论研究却鲜有报道。稀土掺杂会改变半导体材料的能带结构和光学性质[18-23]。鉴于此,本文采用第一性原理赝势平面波方法,对稀土元素La掺杂磷烯的几何结构、态密度和光学性质进行计算,为新型二维材料磷烯在光电材料掺杂改性的实验和理论研究方面提供依据。
1 计算模型和方法
1.1 计算模型
本文使用的计算模型磷烯是从黑磷晶体中剥离出来的,具有层状褶皱结构。黑磷属于正交晶系,空间群为Cmca,晶格常数为a=4.376Å,b=10.478Å,c=3.314Å[24]。对于稀土元素La掺杂磷烯的电子结构及光学性质的计算,选择含有36个P原子的晶胞作为未掺杂模型,再用一个La原子置换磷烯晶胞中的一个P原子,坐标是(0.36,0.60,0.50),从而得到La掺杂磷烯的计算模型,如图1所示。
1.2 计算方法
本文采用的计算方法是基于密度泛函理论(DFT)框架下第一性原理赝势平面波方法,计算方面由CASTEP[25]软件包去完成。首先采用BFGS[26]算法完成对磷烯和掺入稀土元素La后的磷烯晶胞几何结构的优化,进一步得到优化后的稳定结构,接下来在各项都稳定的情况下,计算能带结构、态密度和光学性质。为了确保计算精度,设定平面波截断能E=240eV,能量收敛于1×10-6eV/atom,采用超软赝势[27]来处理离子实与电子间的相互作用,选取广义梯度近似GGA的PBE[28]来处理电子交换关联能,选取各原子的价电子为P的3s23p3、La的5s25p65d16s2,布里渊区积分采用4×3×4的Monkhorst-Pack形式[29]的对称特殊k点方法,FFT网格参数设置为48×48×48。
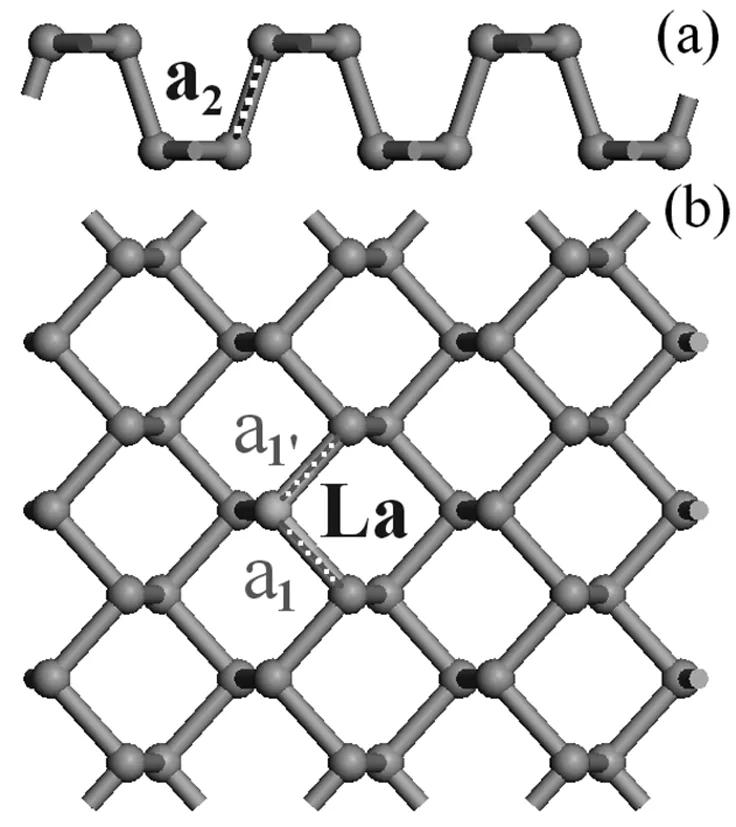
图1 La掺杂磷烯模型 (a)侧视图;(b)俯视图
2 计算结果和讨论
2.1 几何结构
从表1磷烯掺杂前后的键长可以看出:计算采用的磷烯模型在同一平面上的P-P键长均相等(a1= a1’)为2.223Å,与实验值2.224Å的误差不超过0.05%,而不在同一平面上的P-P键长a2的计算值为2.245Å,与实验值2.244Å的误差也是不超过0.05%,说明计算采用的磷烯模型是可靠的。同时,从表1还可以看出,La掺杂后,在掺杂原子附近,与La原子相连的三个P原子的La-P键长发生了明显的变化,如 a1、a1Å、a2都比未掺杂时增大。产生这些变化的原因是由于P和La的原子半径不同造成的,如P的原子半径是1.10Å,而La的原子半径是1.87Å。由于原子半径的差别,在掺杂原子附近引起了畸变,使得磷烯的物理结构发生了变化,说明稀土La掺杂磷烯可以调整其结构参数。

表1 磷烯掺杂前后的键长
2.2 电子结构
图2-图3为La掺杂磷烯前后的能带结构和态密度。通过图2(a)可以看出,未掺杂的磷烯能带结构为直接带隙,并在T点取得,对应带隙宽度为0.923eV。在-15eV-6eV的下价带主要由P的3s态构成;在-6eV~0eV的上价带和0eV~5eV的导带主要由P的3p态构成,由图3(a)可以给出解释。La掺杂磷烯后,能带结构如图2(b)发生了显著的变化。表现在图2(c)中导带底向上平移而价带顶不动,仍在T点取得直接带隙,带隙宽度增大变为1.067eV。同时,价带比未掺杂时增多变密,此时,价带分成三组,最低价带出现在-33.954eV,而且只有一条,这是由La的5s和6s态构成;在能量低于-17.574eV时,出现了由三条能带构成的一组能带,它是由La的5p态构成。在能量高于-17.574eV时的价带,除了P的3s和3p态的贡献之外,还有部分La的5d态的贡献。这些可由图3(b) 给出解释。
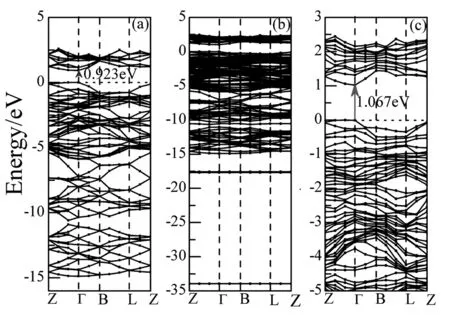
图2 磷烯的能带结构.(a)未掺杂;(b)La掺杂;(c)费米面附近的La掺杂
从图3还可以看出:La掺杂后磷烯的态密度发生了显著的变化。如在费米面附近,电子态密度峰值向低能方向移动且减小。表现如下:在由掺杂前在1.616eV的峰值移到了1.406eV,峰值由33.754减小到23.245;由掺杂前在-1.233eV的峰值移到了-1.286eV,峰值由51.106 减小到37.656。这些是由于少量La的5d轨道和P的3p轨道电子发生杂化减少了带间电子的跃迁造成的。
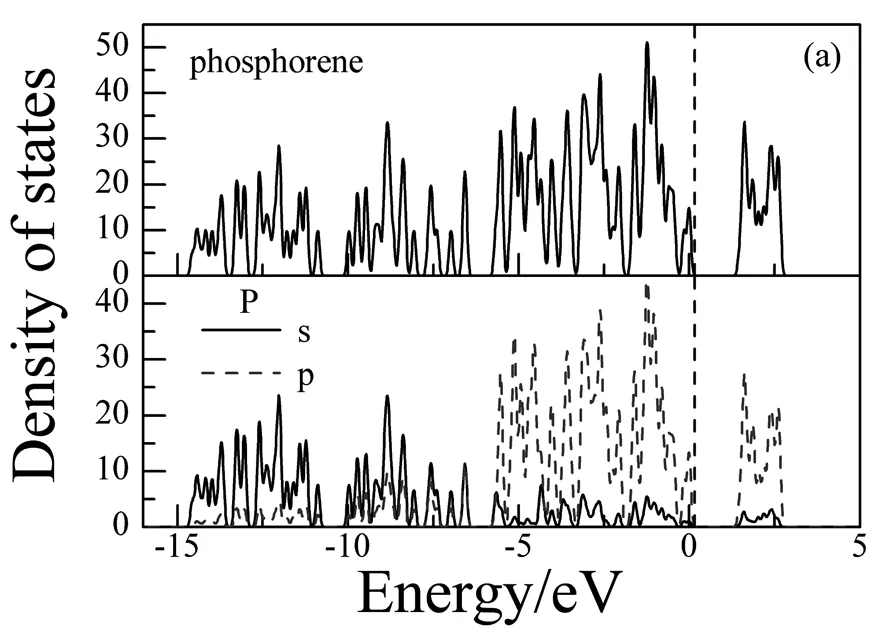
图3磷烯的态密度
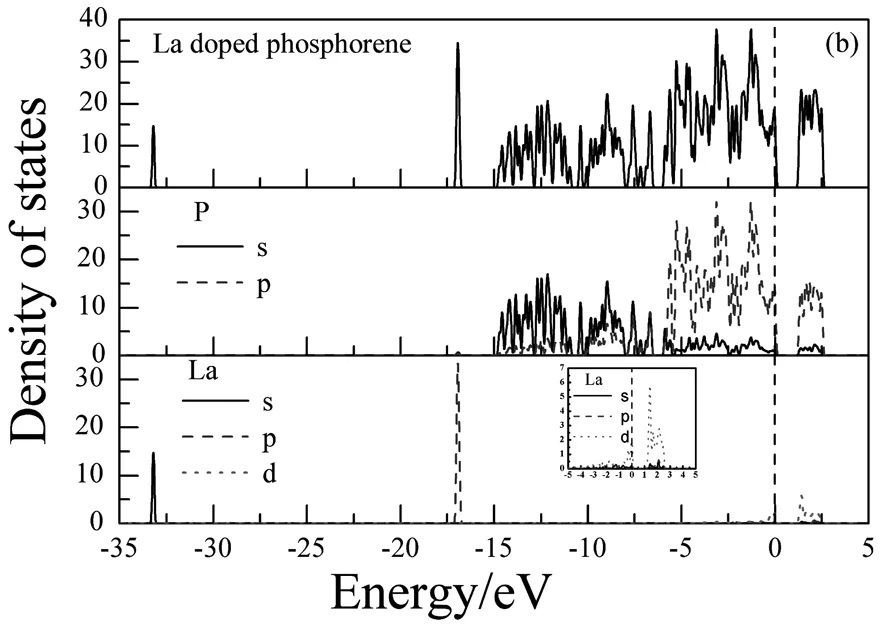
(b) La掺杂
2.3 介电函数
图4为掺杂前后磷烯的介电函数的实部和虚部。通过图4(a)可以看出,磷烯的静态介电常数ε1(0)为1.539,介电常数的虚部ε2在能量低于0.923eV时为零,这与前面计算的带隙宽度一致。与图4 (b)进行对比,可以看出:La掺杂后,介电函数发生了显著的变化。磷烯的静态介电常数ε1(0)增加到3.365,虚部ε2的光学吸收边为1.067eV,对应的介电峰数值明显增大,这是由于La的5d态电子的加入,使光电子跃迁的强度增大。
2.4 吸收系数
由图5可以看出,磷烯的吸收峰在5.756eV处取得,这是由P的3p态电子从价带跃迁至导带产生的。La掺杂后吸收系数发生了显著的变化,在能量大于1.067eV而小于15eV的范围内,吸收峰由掺杂前在5.756eV取得变为在6.191eV取得,吸收峰峰值明显增大。在能量为19.740eV处出现了一个新的吸收峰,这是La的5p态电子从价带跃迁至导带产生的。这些结果说明可以利用La掺杂来提高磷烯的光电转换效率。
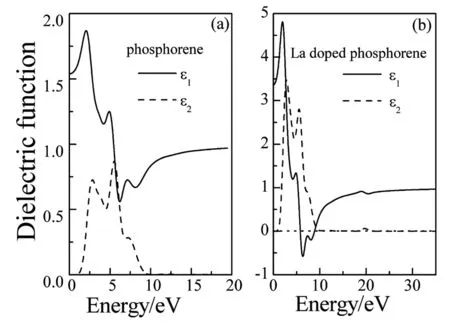
图4 磷烯的介电函数 (a)未掺杂;(b) La掺杂
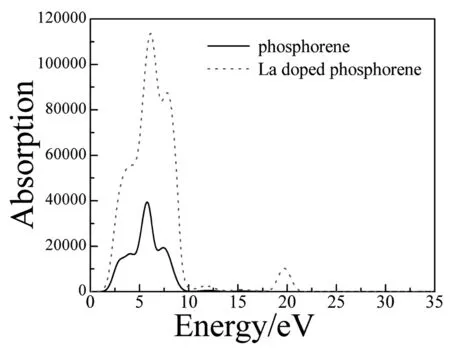
图5 未掺杂和La掺杂磷烯的吸收系数
3 结 论
采用密度泛函理论的第一性原理赝势平面波的方法,对磷烯掺入稀土元素La前后的物理结构、电子结构、介电函数和吸收系数进行了理论计算。计算结果表明:用La原子置换一个P原子后,在掺杂原子附近引起了畸变,使得磷烯的物理结构发生了变化。掺入稀土La后能带结构仍为直接带隙,带隙宽度变宽,带隙宽度由未掺杂时的0.923eV变为1.067eV,能带数目增多,态密度峰值减小。与未掺杂时相比,由于La的5d态电子的加入,使光电子跃迁的强度增大,静态介电常数1(0)增大,虚部2的峰值明显变大,并向低能方向有偏移。同时,吸收系数增大,吸收峰增多,说明可以利用La掺杂来提高磷烯的光电转换效率。希望以上计算结果能为新型二维材料磷烯在光电材料掺杂改性的实验和理论研究方面提供依据。
