掺杂改性对钙铝基复合物酯交换催化剂吸附性能影响的分子模拟
2020-08-19厉志鹏牛胜利韩奎华路春美
厉志鹏,牛胜利,韩奎华,路春美
(山东大学能源与动力工程学院,山东济南250061)
引 言
随着全球范围内气候变暖情况的出现,人们对绿色可持续可再生燃料的关注日益增加。与化石燃料相比较,生物柴油有着无毒、可生物降解、可再生等诸多优势,并且具有生命周期近碳中性优势[1]。在商业生产中,以脂肪酸甲酯(FAME)形式存在的生物柴油是使用均相碱催化剂催化甘油三酸酯(植物脂肪和动物油脂的主要成分)与甲醇的酯交换反应生产的。尽管该催化过程在温和的条件下便能提供很高的反应速率,但甘油三酸酯与甲醇进行酯交换反应时,将产生大量副产物甘油,产物净化程序复杂,由此增加了生物柴油的生产成本[2],采用乙酸甲酯代替甘油三酸酯与甲醇进行反应,酯交换生成的副产物为甘油三乙酸酯,可完全溶于脂肪酸甲酯,作为燃料生物添加剂成为生物柴油燃料的一部分,同时这也是当前分子模拟普遍采用的方法[3-4]。该生产过程的另一个缺点就是均相催化过程催化剂分离困难,并产生大量废水,相较而言,非均相催化是一种环境友好的生产生物柴油的有效途径。
γ-Al2O3具有极高的热稳定性、高机械稳定性、高比表面积以及较大的孔径和孔容,作为催化剂载体已经广泛应用于多种化学反应过程,如聚合[5]、改性[6]、脱水[7]、质子化[8]。另外,钙基催化剂碱性强,催化酯交换反应条件温和。目前,已有大量关于单一钙基[9]及其复合金属化合物[10]催化甘油三酸酯与甲醇酯交换反应生成生物柴油的相关研究。但是,钙铝二元复合氧化物固体碱催化剂仍然存在比表面积较小导致的催化活性弱以及活性位浸出率较高造成的重复使用性差等问题。Zheng 等[11]制备的钙铝多相催化剂在6 次循环使用后催化剂的质量从0.150 g 减少到0.075 g,活性位氧化钙损失较多导致失活。Tang 等[12]在对钙铝复合催化剂进行SEM 分析时指出,钙铝复合催化剂颗粒较大,团聚现象严重,不利于酯交换反应的进行。因此,目前研究的热点主要集中在提高钙基催化活性以及降低钙基活性位流失率等方面。
锌、镧、镁改性可有效强化CaO 的催化酯交换性能。锌-钙氧化物共存可以提高催化剂的比表面积和比孔容,形成粒径小的钙锌复合物,提高催化酯交换的活性[13]。La3+拥有完整的4d和空白的5d轨道以提供或接收电子,使催化剂拥有酸碱双性位,其键合作用能有效抑制催化酯交换过程中钙基活性位溢出,使催化剂具有较高的重复使用性[14]。与纯CaO 催化剂相比,镁改性的钙镁复合物催化剂可降低活性位浸出率、提高催化剂稳定性、延长催化剂的使用寿命[15]。相关研究同时得出,分别以Al2O3和CaO 作为催化剂的载体和活性位,进一步进行锌[16]、镧[17]或者镁[18]的掺杂改性,能显著提高铝钙复合物的催化性能,比单纯的钙铝复合催化剂效果更佳,但其中的作用机制尚不清楚。
本文基于密度泛函理论(density function theory,DFT),对Ca-Al-M(M=Zn,La,Mg)复合物催化剂表面的电子结构特性进行了分子模拟,计算了复合催化剂形成时的掺杂结合能,系统地探究了甲醇在催化剂表面的吸附情况,通过Mulliken 电荷布局分析讨论了甲醇在催化剂表面吸附后电荷的变化,在分子层面上解析了锌、镧、镁掺杂改性对钙铝复合物催化剂催化酯交换过程的影响机制,所得结论可为钙铝基固体碱酯交换催化剂的设计与开发提供理论指导。
1 计算方法和模型
1.1 计算方法
本研究基于密度泛函理论,采用自旋非限制广义 梯 度(generalized gradient approximation)耦 合Perdew-Wang 91 泛函的方法(GGA-PW91)在DMol3水平上完成[19]。选用有效核心赝势(DFT semi-core pseudopod)以降低计算成本,同时价电子波函数采用双数值基加轨道极化函数(DNP)展开,并将Monkhorst-Pack 网格参数k 设置为2×2×1。计算精度为Customized,自洽场最大迭代次数为300,选用热拖尾效应(smearing),并保持初始默认值0.136 eV。结构优化以梯度、位移和能量是否收敛为判据,最大梯度、最大位移和最大能量的收敛精度均分别优于0.110 eV、5.0×10-4nm 和5.4×10-3eV/nm,同时,SCF收敛判据的截断值tolerance为1.0×10-5。
本文计算了酯交换反应物甲醇、乙酸甲酯分子的键长,并与文献结果对比以确保优化方法的准确性。甲醇和乙酸甲酯的结构如图1 所示,计算结果见表1,其中键长与文献[4,20]结果相符,因此本文所用计算方法可行。

表1 甲醇和乙酸甲酯的键长Table 1 Bond length of methanol and methyl acetate
1.2 模型构建
本文所用γ-Al2O3结构的晶体数据来自Digne模型[21]。对该结构进行进一步构型优化以完善模型,计算得出γ-Al2O3晶体参数分别为a=0.559 nm、b=0.841 nm、c=0.807 nm、β=9.059 nm,与文献结果吻合。鉴于(110)表面为γ-Al2O3晶体暴露最丰富表面[22],因此本文选用γ-Al2O3(110)表面作为计算表面。模型采用p(1×1)的7 层周期性平板,并在平板与真空层构成超单元晶胞,同时在z 方向构建1 nm真空区以避免连续平板之间的相互作用,从而在空间中周期性重复,最终得到γ-Al2O3(110)表面模型,如图2 所示。考虑表面弛豫影响,固定底部四层原子,同时保持上部三层及吸附分子自由移动。γ-Al2O3(110)表面暴露有三配位铝(Al3c)、四配位铝(Al4c)、五配位铝(Al5c)以及二配位氧(O2c)、三配位氧(O3c),其中三配位铝(Al3c)、四配位铝(Al4c)、二配位氧(O2c)未饱和,这与Song等[23]的研究结果一致。
氧化铝表面的掺杂结合能(Edope)(单原子掺杂)由式(1)计算。

氧化铝表面的掺杂结合能(Edope)(双原子掺杂)由式(2)计算。
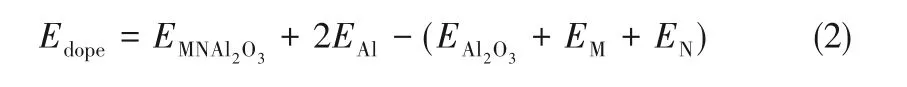
催化剂表面的吸附能(Eads)由式(3)计算。吸附能可以用来衡量吸附体系的稳定性,其值越大表明吸附过程中放出热量越多,吸附越稳定,催化剂催化效果越好。
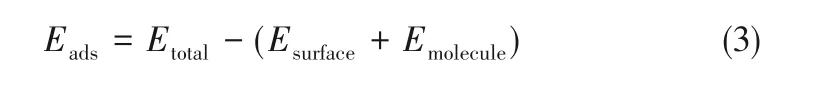
2 结果和讨论
2.1 Ca-Al-M复合催化剂的构成
在对Ca-M 在氧化铝表面掺杂过程中,考虑到γ-Al2O3(110)表面有三个不同配位的铝原子(Al3c-1,

图1 甲醇结构和乙酸甲酯结构(红色为氧原子,白色为氢原子,灰色为碳原子)Fig.1 Structure of methanol and methyl acetate(O atoms are red,H atoms are white and C atoms are gray)

图2 γ-Al2O3(110)结构表面(红色为氧原子,粉色为铝原子)Fig.2 Structure of the γ-Al2O3(110)surface(O atoms are red and Al atoms are pink)
Al4c-1,Al5c-1),利用原子替换法[24]分别对氧化铝进行掺杂,掺杂的类型及优化后掺杂结合能如表2 所示。掺杂结合能为正值表明该掺杂过程不能自发进行,相应的值越小,表明掺杂过程越容易进行,掺杂得到该结构的可能性越大[25]。结果表明,Ca-Al 复合物催化剂中的钙原子倾向于掺杂在五配位铝Al5c的位置;Ca-Al-Zn 复合物催化剂中的钙原子、锌原子分别倾向于掺杂在四配位铝Al4c、五配位铝Al5c的位置;Ca-Al-La 复合物催化剂中的钙原子、镧原子分别倾向于掺杂在三配位铝Al3c、五配位铝Al5c的位置;Ca-Al-Mg 复合物催化剂中的钙原子、镁原子分别倾向于掺杂在五配位铝Al5c、三配位铝Al3c的位置。相应地,在其最易掺杂位置的掺杂结合能分别为214.50、893.98、151.23 以及643.25 kJ/mol,同时,掺杂后的催化剂构型如图3所示,其中,当钙、钙锌、钙镧、钙镁在氧化铝表面掺杂的时候,五配位铝Al5c最易于被掺杂,与杨涛等[26]的研究结果一致。

表2 催化剂表面在不同情况下的掺杂结合能Table 2 Dopant incorporation energy of catalysts surface under different conditions

图3 掺杂改性后催化剂表面结构俯视图(红色为氧原子,粉色为铝原子,绿色为钙原子,黄色为镁原子,黑色为锌原子,蓝色为镧原子)Fig.3 Top view of doped catalyst structure surfaces(O atoms are red,Al atoms are pink,Ca atoms are green,Mg atoms are yellow,Zn atoms are black and La atoms are blue)
2.2 反应物在Ca-M催化剂上的吸附
在Ca-Al-M(M= Zn, Mg, La)三元复合物催化剂催化酯交换反应过程中,由于γ-Al2O3主要起催化载体的作用,为使锌、镁、镧等掺杂改性剂对钙基活性位的调节作用更加明显,在本节将对酯交换反应物在Ca-M 二元复合物催化剂上的吸附进行讨论。在酯交换生产生物柴油的过程中,甲醇和乙酸甲酯首先吸附在催化剂表面,其中甲醇在催化剂的作用下发生解离,生成甲氧基和氢离子,甲氧基进一步攻击乙酸甲酯上的碳氧双键,生成脂肪酸烷基类酯,即生物柴油[27]。本节将主要讨论甲醇和乙酸甲酯在催化剂表面吸附过程中,催化剂对甲醇上氢氧键及乙酸甲酯上碳氧双键的影响。
分别构建甲醇在钙基催化剂以及钙锌、钙镧、钙镁二元复合物催化剂上吸附的模型,构型优化如图4 所示。甲醇在催化剂表面吸附时,氢氧键发生解离,吸附后甲醇的H—O 键长度、催化剂表面H—M 键长度以及吸附能如表3 所示。吸附能可以用来衡量吸附体系的稳定性,其值越大表明吸附过程中放出热量越多,吸附越稳定。根据表3 可以看出,锌、镁掺杂改性剂能显著提高甲醇在催化剂表面的吸附能,使得反应释放更多能量,且H—O 键长度较吸附前(表1)明显增加,同时H—M 键长度明显减小,说明解离产生的氢原子与催化剂表面的氧原子形成新的化学键,最终强化了甲醇的吸附效果及活性程度;采用镧作为掺杂改性剂时,吸附能有略微减少,但H—O 键长度较吸附前明显增加,且H—M键长度明显减小,也在一定程度上增加了甲醇的活化程度。
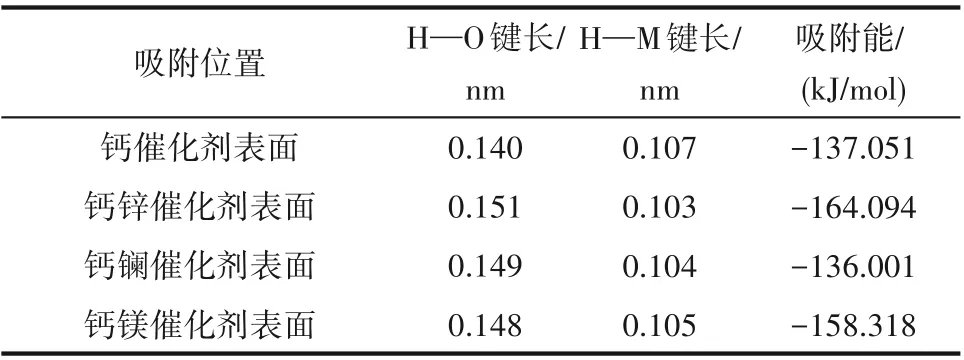
表3 甲醇吸附在催化剂表面后的键长和吸附能Table 3 Bond length and adsorption energy of methanol absorbed on catalyst surfaces

图4 甲醇分子在Ca-M催化剂表面上的吸附构型Fig.4 Structure of methanol molecule absorbed on Ca-M catalyst surfaces
分别构建乙酸甲酯在钙基催化剂以及钙锌、钙镧、钙镁二元复合物催化剂上的吸附模型并进行构型优化。吸附后,乙酸甲酯碳氧双键C O 的键长变化及吸附能见表4。与催化剂对甲醇吸附结构的影响相比,催化剂对乙酸甲酯的碳氧双键(表1)的作用微乎其微。当采用锌、镧作为掺杂改性剂时,吸附能明显增大,有利于催化酯交换反应的进行;当采用镁作为掺杂改性剂时,吸附能反而减小,表明反应发生的难度增加,镁的添加不利于酯交换反应的进行。

表4 乙酸甲酯吸附在Ca-M催化剂表面后的键长和吸附能Table 4 Bond length and adsorption energy of methyl acetate absorbed on Ca-M catalyst surfaces
由表3、表4 可知,反应物在单一的钙基催化剂表面以及在钙锌、钙镧、钙镁的二元复合物催化剂表面的吸附能分别为-257.691、-299.196、-285.695、-259.119 kJ/mol,即采用锌、镧、镁作为掺杂改性剂均能增大反应物的吸附能,且活化甲醇的能力均得到增强,有利于催化酯交换反应的进行。付希[28]指出,采用溶胶-凝胶法制备的CaO-ZnO 固体碱催化剂具有较大的孔隙,能在保证催化效果的前提下具有优异的稳定性;Yan 等[29]通过多种制备方法研究出的钙镧二元复合催化剂拥有更高的BET 比表面积,浓度更高的强碱位点,在酯交换反应中拥有极高的催化活性;王广欣等[30]的研究表明CaO/MgO 催化剂沉降性好,抗中毒能力强,浸出率低,相较氧化钙催化性能得到较大改善;与计算结果相符。
2.3 甲醇在Ca-Al-M催化剂上的吸附
2.3.1 甲醇在Ca-Al-M 催化剂上的吸附能及吸附构型 根据图3 可知,由于γ-Al2O3(110)有较高的比表面积,孔隙较大,当采用多种原子对其掺杂后,仍然保持着优异的物理结构,有利于增大醇油分子的吸附效率[31]。根据2.2 节甲醇、乙酸甲酯在Ca-M 二元复合物催化剂表面的吸附结果,采用锌、镧、镁对钙进行掺杂改性均能增强催化剂的催化效果,且甲醇在催化剂表面吸附时催化剂对其氢氧键活化效果最为显著,为催化剂的主要作用部分。因此,本节将只考虑甲醇在Ca-Al-M 三元复合物催化剂表面的吸附,吸附构型如图5 所示,吸附后甲醇中H—O键长度和吸附能如表5所示。
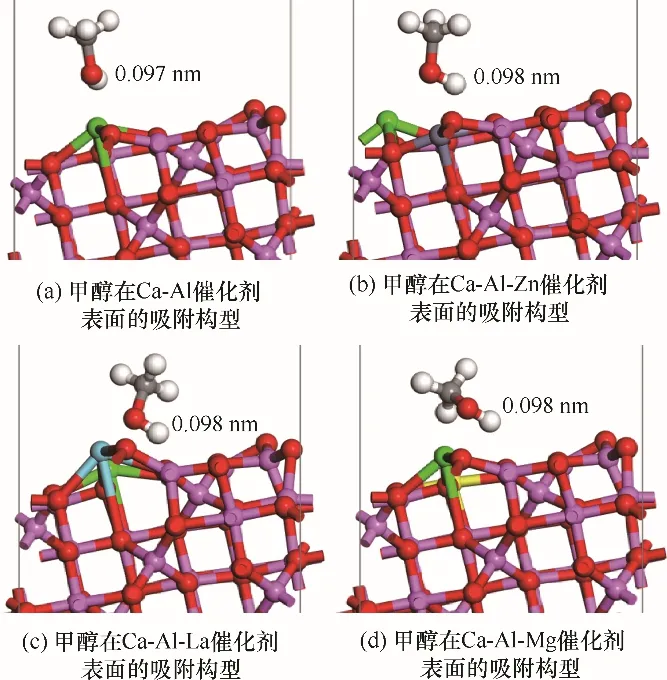
图5 甲醇分子在Ca-Al-M 催化剂表面的吸附Fig.5 Structure of methanol molecule absorbed on Ca-Al-M catalyst surfaces
根据表5 可知,在采用锌、镧、镁对钙铝基催化剂掺杂改性后,甲醇在催化剂表面的活化效果均得到增强,且吸附能均增大,在γ-Al2O3载体作用下,镧掺杂改性时甲醇吸附能略微减小的缺点已经克服,催化剂催化效果变强。Norhasyimah 等[32]指出,具有三倍氧化铝涂层的单载体氧化钙催化剂在添加锌物质的情况下生物柴油的转化率显著提高;Syamsuddin 等[33]在钙铝比为6∶1 的钙铝二元复合催化剂中添加镧至钙镧铝比为6∶2∶1 后,FAME 收率由80%上升到了96.91%;Gao 等[34]的实验结果表明,在KF 的作用下,钙铝二元复合催化剂催化酯交换反应1 h 后FAME 收率可达96.6%,而钙镁铝三元复合催化剂作用10 min 后FAME 收率便可达98%,与本文分析规律一致。
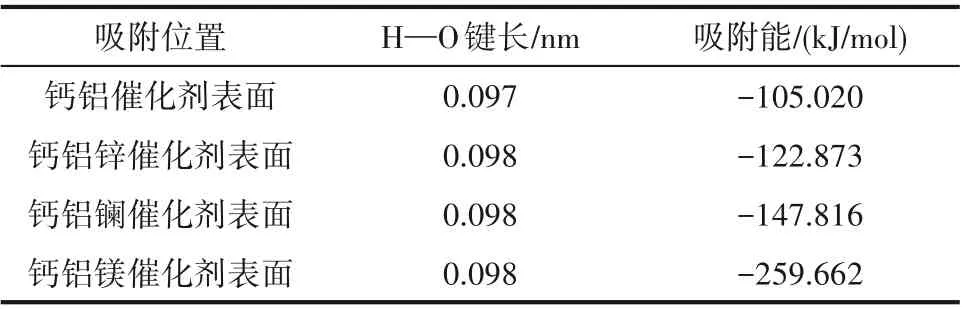
表5 甲醇吸附在Ca-Al-M 催化剂表面后的键长和吸附能Table 5 Bond length and adsorption energy of methanol absorbed on Ca-Al-M catalyst surfaces
2.3.2 甲醇在Ca-Al-M 催化剂上吸附的Mulliken电荷密度分析 Mulliken 电荷布局分析[35]反映吸附体系中原子与键在吸附前后的电子分布变化。如表6所示,吸附前后甲醇上羟基中氢原子电正性和氧原子电负性均发生变化,说明在催化剂的作用下,甲醇的电荷进行了重新分配。采用锌作为调节剂时,氢原子电正性和氧原子电负性分别增大了0.052、0.022 e,说明甲醇得到了更好的活化;采用镧作为调节剂时,氢原子电正性和氧原子电负性分别增大了0.078、0.034 e,也说明甲醇得到了更好的活化;采用镁作为调节剂时,氢原子的电正性增大了0.026 e,氧原子的电负性减小了0.005 e,但增大数值明显大于减小数值,仍说明甲醇得到了更好的活化。将甲醇上各个原子电荷密度相加得到甲醇分子总电荷密度的变化值,说明甲醇在催化剂表面吸附之后失去电子,且相较于钙铝Ca-Al二元复合物催化剂,使用锌、镧、镁掺杂改性后,Ca-Al-M 三元复合物催化剂失去电子的数量增加,采用锌、镧、镁进行调节后,甲醇失电子数分别增加0.016 e、0.044 e、0.006 e。以上分析表明,甲醇分子吸附到催化剂表面时,甲醇与催化剂表面活性中心之间存在电子传递过程,从而增强了甲醇分子的吸附稳定性,同时也活化了甲醇分子,进一步掺杂改性后,甲醇与催化剂表面的电子转移数增加,说明甲醇的活化程度增强[36]。结果表明,采用锌、镧、镁作为调节剂时,更有利于吸附反应的发生,甲醇的活化得到增强,这与吸附能和结构分析的结果一致。
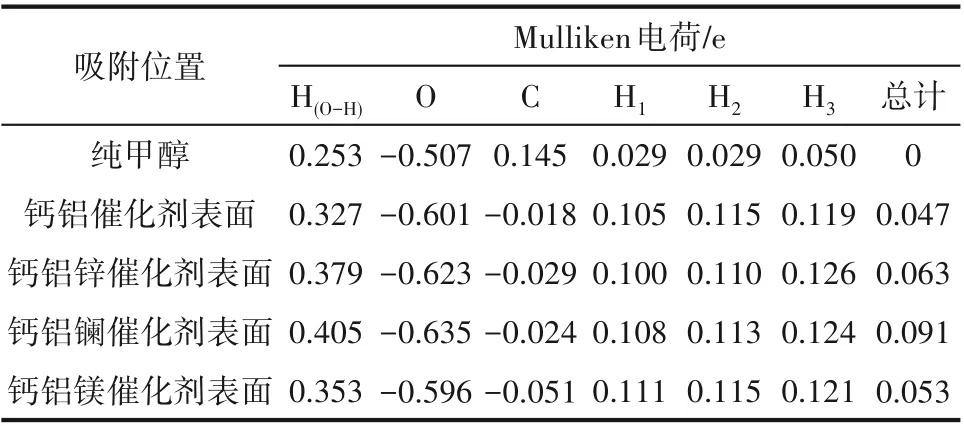
表6 甲醇吸附在Ca-Al-M催化剂表面的Mulliken电荷布局Table 6 Mulliken atomic charge populations for methanol adsorption on Ca-Al-M catalyst surfaces
3 结 论
(1)分别优化了钙铝、钙铝锌、钙铝镧、钙铝镁复合催化剂的几何构型,结果表明钙铝二元复合催化剂中钙原子倾向于掺杂在五配位铝的位置,钙铝锌三元复合催化剂中钙原子、锌原子分别倾向于掺杂在四配位铝、五配位铝的位置,钙铝镧三元复合催化剂中钙原子、镧原子分别倾向于掺杂在三配位铝、五配位铝的位置,钙铝镁三元复合催化剂中钙原子、镁原子分别倾向于掺杂在五配位铝、三配位铝的位置。
(2)锌、镧、镁对钙进行掺杂改性能有效地增强其对甲醇的活化能力,当反应物在催化剂表面吸附时,吸附能均增大,其中甲醇在催化剂表面的吸附是酯交换反应的决速步骤。
(3)γ-Al2O3凭借其优异的结构特征,作为催化反应的载体能有效增强催化效果。甲醇在钙铝二元复合催化剂上吸附时,采用锌、镧、镁进行掺杂改性后,甲醇在催化剂表面的吸附能明显增大,甲醇活化程度增强。通过对甲醇进行Mulliken电荷密度分析,进一步验证了锌、镧、镁增强催化剂催化能力的效果。
符 号 说 明
EAl——Al单原子能量,kJ/mol
EAl2O3——氧化铝表面能,kJ/mol
Eads——吸附能,kJ/mol
Edope——掺杂结合能,kJ/mol
EM——M单原子能量,kJ/mol
EMAl2O3——M原子掺杂Al2O3表面后的总能量,kJ/mol
EMNAl2O3——M 和N 原子掺杂Al2O3表面后的总能量,kJ/mol
Emolecule——分子吸附前的总能量,kJ/mol
EN——N单原子能量,kJ/mol
Esurface——催化剂表面能,kJ/mol
Etotal——催化剂表面吸附分子后的总能量,kJ/mol
