Waardenburg综合征II型SOX10基因突变一家系分析并文献综述
2020-08-08韩瑞钰邓佩佩张文昊王树松刘效群
韩瑞钰 邓佩佩 闫 蒙 张文昊 王树松 刘效群
河北省计划生育科学技术研究院(河北省生殖医学中心), 国家卫生健康委计划生育与优生重点实验室/河北省生殖医学重点实验室(石家庄,050071)
Waardenburg综合征(WS)又称听力-色素综合征,由荷兰眼科学家Waardenburg在1951年首先提出,是一种较为常见的综合征遗传性耳聋,占先天性耳聋的2%~5%,其发病在男女及种族间无明显差异[1-2]。临床表现以先天性感觉神经性听力丧失及虹膜、头发和皮肤的色素异常沉着为主要特征,目前认为其病因主要是由神经嵴细胞发育缺陷或障碍引起。根据临床表现的不同分为Ⅳ型,主要致病基因包括:EDN3,EDNRB,MITF,PAX3,SNAI2,SOX10等[3]。研究报道显示WSⅠ、WSⅡ、WSⅢ型为常染色体显性遗传,WSⅣ型中由SOX10基因突变引起的为常染色体显性遗传,由EDN3和ED-NRB基因突变导致为常染色体隐性遗传。本研究报道了1例SOX10新发基因突变导致WSⅡ型的家系病例。
1 材料与方法
1.1 临床资料
先证者及其父母、哥哥来自中国河北省沧州地区,询问先证者及其父母、哥哥的临床表现,取得家庭成员的知情并签署知情同意书后获取先证者及其父母的外周静脉血各5ml,用于基因组DNA提取。
1.2 检测方法及突变验证
提取先证者及其父母、哥哥全基因组DNA,建立DNA文库,在Illumina HiSeq X10平台进行全基因组重测序,测序数据首先运用BWA软件与人类参考基因组GRCh37/hg19参考序列进行比对,再用GATK软件识别基因组SNP、INDEL、CNV突变位点,结果参考数据库HGMD Pro、PubMed、1000Genomes及 dbSNP,发现突变位点采用Sanger测序方法进行验证。
2 结果
2.1 临床资料分析
先证者,女(图1,Ⅱ2),30岁,先天性耳聋,右侧眼睛虹膜异色,呈宝石蓝色,无其它临床症状;母亲(图1,Ⅰ1),先天性耳聋,可发出声音,丧失语言能力,双侧眼睛虹膜异色,呈蓝色,早年额前白发,无其它临床症状;父亲与哥哥表现正常,除先证者及其母亲外均无此症状。家系图见图1。
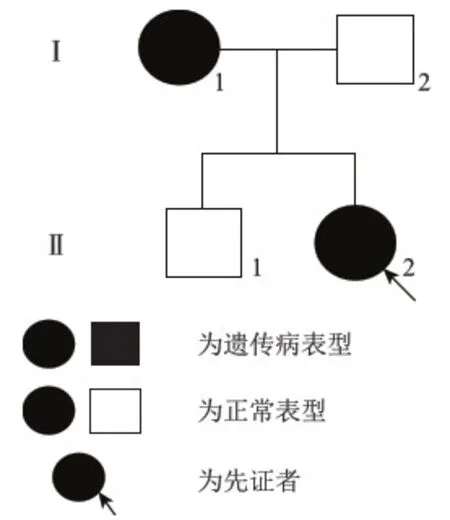
图1 Waardenburg综合征Ⅱ型家系图
2.2 突变分析
基因检测,在先证者的SOX10基因发现c.425G>A杂合核苷酸变异,编码区第425号核苷酸由G变为A,该变异导致编码第142号氨基酸Trp的密码子变为终止密码子,从而使肽链合成提前终止,为无义突变(图2A)。母亲基因检测结果与先证者一致,该变异可能导致蛋白质功能受到影响,但致病性尚未见报道(参考数据库:HGMD Pro及PubMed),其不属于多态性变化,在人群中的发生的频率极低(参考数据库:1000Genomes、dbSNP)(图2C)。父亲与哥哥基因检测未发现基因突变位点(图2B、D)。根据临床症状及基因突变结果分析该先证者为Waardenburg综合征Ⅱ型。测序结果见图2。
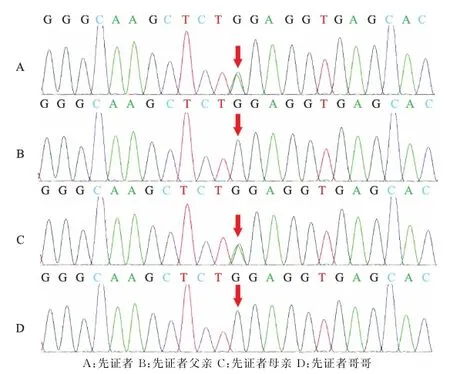
A:先证者 B:先证者父亲 C:先证者母亲 D:先证者哥哥
2.3 WSⅡ型SOX10基因突变文献回顾与总结
查阅文献报道,总结分析发现引起WSⅡ型SOX10基因突变共有32种(表1),包括18例缺失突变,2例插入突变,11例点突变,1例合并缺失与插入突变。从表中可以发现突变多发生于男性,30例突变患者中,有6例女性,5例没有标明性别,21例为男性。且在中国人群中发现报道较多,26例均为中国患者,占81.25%,除1例突变有报道外,其余均为新发突变,32例病例中有5例明确的家族遗传倾向。
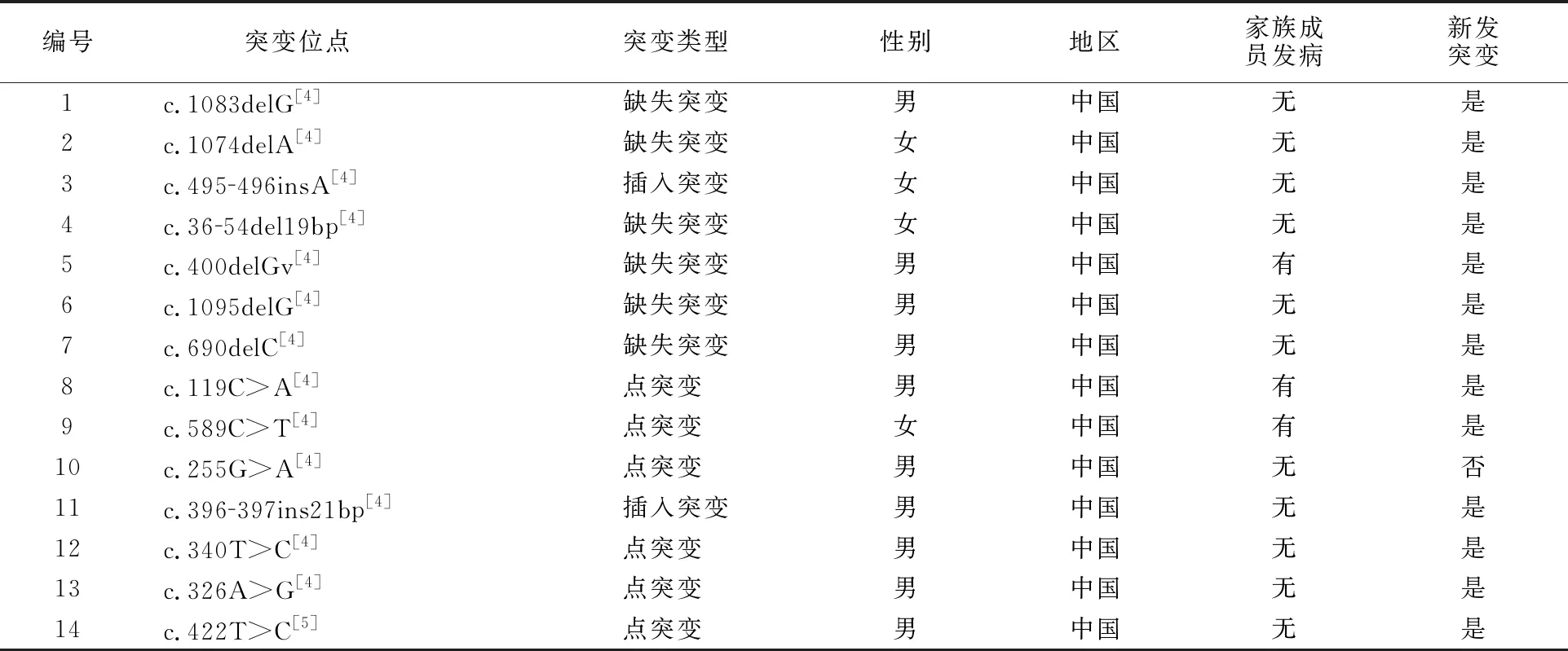
表1 已报道的WSⅡ型患者的SOX10基因突变总结
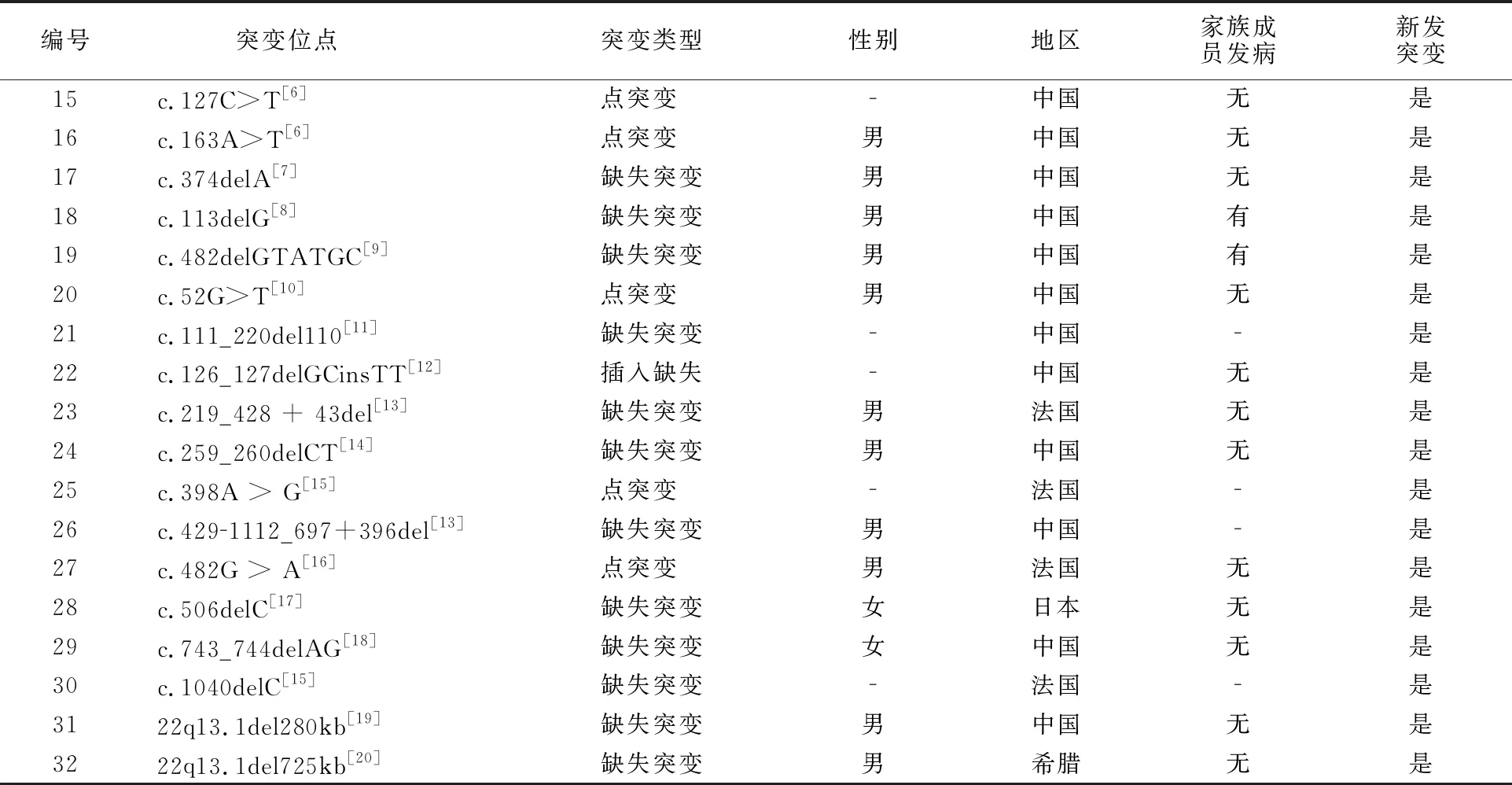
编号突变位点 突变类型性别地区家族成员发病新发突变15c.127C>T[6]点突变 中国无是16c.163A>T[6]点突变 男中国无是17c.374delA[7]缺失突变男中国无是18c.113delG[8]缺失突变男中国有是19c.482delGTATGC[9]缺失突变男中国有是20c.52G>T[10]点突变 男中国无是21c.111_220del110[11]缺失突变中国是22c.126_127delGCinsTT[12]插入缺失中国无是23c.219_428 + 43del[13]缺失突变男法国无是24c.259_260delCT[14]缺失突变男中国无是25c.398A > G[15]点突变 法国是26c.4291112_697+396del[13]缺失突变男中国是27c.482G > A[16]点突变 男法国无是28c.506delC[17]缺失突变女日本无是29c.743_744delAG[18]缺失突变女中国无是30c.1040delC[15]缺失突变法国是3122q13.1del280kb[19]缺失突变男中国无是3222q13.1del725kb[20]缺失突变男希腊无是
2.4 生育风险评估
本研究先证者与母亲的突变位点一致,该突变应为先证者母亲新发的致病性突变,先证者的基因突变是由母亲遗传而来,鉴于该疾病为常染色体显性遗传病,且母亲与先证者均为杂合突变,对于先证者生育的遗传咨询其生育患儿的几率为50%,建议生育时行产前诊断,或辅助生殖技术避免患儿出生。
3 讨论
SOX10基因位于染色体22q13.1,含有5个外显子,其中只有3、4、5号外显子编码蛋白,5’端非编码区含有1个或1个以上的非编码外显子,SOX10蛋白含有466个氨基酸,主要功能是识别并结合靶基因启动子DNA[21]。目前研究认为 SOX10基因是神经嵴发育的关键调节基因,如果发生突变会导致神经嵴性疾病,如以耳聋及色素异常为主要表型的 Waardenburg综合征,有研究发现SOX10在内耳发育早期有广泛表达[22-23]。本研究中先证者及其母亲发现SOX10基因c.425G>A的杂合核苷酸变异,为无义突变,SOX10基因突变主要引起WSⅡ型和WSⅣ型,40%~45%WSⅣ型由SOX10基因变异引起,15%的WSⅡ型由SOX10基因变异引起,且WS患者检测到的SOX10基因突变绝大多数为终止密码子提前出现,从而产生截短蛋白的框码移位突变及无义突变[24]。
基因突变类型从表1中看出是由SOX10基因突变引起的WSⅡ型,主要表现为缺失突变。2007年Bondurand等[13]发现了第一例SOX10缺失突变导致的WSⅡ型,并提出SOX10基因缺失突变会引起的单倍体剂量不足效应,从而影响了蛋白功能。Siomou等[20]也发现了此效应引起的WSⅡ型综合征。SOX10基因编码的是一种转录因子,可以结合靶基因启动子DNA,研究认为SOX10可与MITF启动子特异结合提高MITF的转录活性,与PAX3协同作用可增强这种激活效应,从而保证神经嵴的发育。根据Wang等[5]研究得知SOX10错义突变c.422T>C,使SOX10蛋白丧失了DNA结合能力,因而不能与PAX3相互作用,最终不能有效激活MITF启动子,抑制黑色素合成而引起WSⅡ型。本研究病例中发现SOX10基因变异出现了WSⅡ型综合征的症状和遗传模式,因此我们推断此病例的基因变异也是因为最终不能激活MITF启动子,抑制黑色素合成而引起相应的临床症状。Zhang等[25]也探讨SOX10基因突变E248fs致Waardenburg综合征Ⅱ型的分子机制,结果表明E248fs突变蛋白完全失去调控MITF启动子转录活性作用,影响了靶基因 MITF 转录活性,导致黑色素合成减少,最终因单倍体剂量不足效应致WSⅡ型。
本研究的突变位点为一新的治病突变基因,可以丰富基因突变谱。本文通过对WSⅡ型SOX10基因突变的文献报道进行回顾和总结,发现引起WS的新发突变较多。 Sun等[4]研究了36名中国汉族耳聋先证者和另外16名临床诊断为WSⅠ型和WSⅡ型的家族成员的WS的分子病因学和基因型-表型相关性。在29个WSⅡ型先证者中,13个被鉴定为SOX10基因突变。在随后此遗传病的研究中可以增加对该家系中其他成员的检测分析并与正常人群对照,进一步确定该疾病的基因突变位点,也为该家系成员的遗传咨询提供合理指导,此综合征的鉴定和确诊为优生指导提供临床依据。
