Co-P@C催化剂的合成及其加氢脱氧性能
2020-07-08张亮亮陈勇强
张亮亮,陈勇强
(晋中学院化学化工学院,山西晋中030619)
催化在人类文明进步与世界经济发展中扮演着非常重要的角色.它能够以一种高效、绿色和经济的方式将原材料转变为具有高附加值的化工产品和燃料等,因而被广泛应用于能源、化工、食品、医药、电子等各个领域.目前,全世界90%以上的化学生产过程都离不开催化.毫不夸张地说,催化领域的每一次重大突破,都极大地改变了人类的生产与生活方式[1].苯酞作为精细化工产品生产中的重要原料和反应中间体,在有机合成中有着广泛的应用.目前工业上多采用邻苯二甲酰亚胺与NaOH反应或者苯酐还原的方法生产苯酞[2].通过这两种方法获得的苯酞的收率较低,且生产过程中造成了严重的环境污染,不符合现代化工的要求.通过催化苯酐选择加氢脱氧合成苯酞是一条理想的生产苯酞的方法.因此,开发高效的苯酐加氢脱氧制备苯酞的催化剂受到了广大学者的关注.
Au、Ag和Ru等贵金属在苯酐加氢制备苯酞的反应中显示出良好的催化活性[2].2009年,刘迎新教授课题组制备了TiO2负载的Au催化剂,并考察其在苯酐加氢制备苯酞的反应中的催化活性.研究发现Au的最佳担载量为2~3 wt.%,其中2 wt.%Au/TiO2催化剂在180℃和3 MPa的优化反应条件下,苯酐的转化率为94.5%,苯酞的选择性为94.4%[3].随后,刘迎新教授进一步制备了不同氧化物掺杂的TiO2复合氧化物载体负载Au催化剂,其中Au/FeTiO2在180℃和3 MPa的优化反应条件下,苯酐的转化率为97.6%,苯酞的选择性为95.2%[4].Henkelmann等人研究发现,贵金属Ag在苯酐加氢制备苯酞的反应中对苯酞的选择性高达89%[5].虽然贵金属具有优异的催化性能,但是其昂贵的价格和有限的资源限制了它的工业化应用.Larchar等使用Ni催化剂催化苯酐加氢制备苯酞的收率为71.7%[6].Funten等人制备了12 wt.%Ni基催化剂并用于苯酐加氢反应中,苯酞的收率为89.5%[7].刘迎新教授课题组制备了Ni/TiO2-SiO2催化剂,在180℃和3 MPa的优化反应条件下,苯酐的转化率为100%,苯酞的选择性80.4%[8~9].但是,苯酐加氢制备苯酞是一个酸性体系,单纯的Ni基催化剂在该酸性体系易造成金属流失导致稳定性下降.因此,提高催化剂的耐酸性具有重要的意义.磷化镍作为新型催化剂已被广泛地应用在多个催化领域中,但其在催化苯酐加氢脱氧反应方面则几乎没有[10].
有鉴于此,本论文以含钴的金属有机骨架材料UTSA-16为金属源,次亚磷酸钠为磷源,采用程序升温法制备碳包覆的磷化钴催化剂,采用XRD表征催化剂的组成和结构,以苯酐的加氢脱氧反应为探针反应,研究其催化性能,预期开发一种新型的磷化钴催化剂.
1 实验部分
1.1 原料和试剂
四水合乙酸钴、次亚磷酸钠、氢氧化钾、柠檬酸、苯酐,分析纯,上海阿拉丁生化科技股份有限公司;无水乙醇、环己烷,分析纯,天津市科密欧化学试剂有限公司;γ-丁内酯,分析纯,上海阿达玛斯试剂有限公司;去离子水(自制).
1.2 催化剂制备
第一步,采用水热法制备了金属有机骨架材料UTSA-16[11~12].首先,将1.245 g四水合乙酸钴、1.051g柠檬酸、0.842g氢氧化钾放入烧杯中;其次,分别取40mL乙醇和40mL去离子水加入上述烧杯中,超声溶解15min后,将混合溶液转移到水热釜(100 mL)中;最后,水热釜在120℃烘箱中处理24h,过滤后的沉淀物分别用乙醇清洗3次,随后在60℃下真空干燥12h.第二步,取0.5g制备好的UTSA-16样品在氮气中400℃热解4h后获得Co@C.第三步,取0.1g Co@C样品与次亚磷酸钠研磨混合10min,将混合后的样品放管式炉中,在氮气气氛下450℃磷化处理2h,得到样品Co-P@C.
1.3 催化剂表征
催化剂样品的XRD表征采用Shimadzu XRD-6100型X射线衍射仪进行.采用PerkinElme STA-6000V型热重分析仪测试UTSA-16样品随温度变化而发生的重量变化.
1.4 催化剂活性评价
通过苯酐的加氢脱氧反应测试Co-P@C的催化性能.催化反应在高压反应釜中进行,催化剂用量为0.1g,Co-P@C催化剂在H2气氛和400℃下还原2 h.反应条件:含5 wt.%苯酐的反应液,环己烷为内标,γ-丁内酯为溶剂.4.0MPa H2,220°C,转速为700 rpm,产品通过气相色谱仪(天美GC7900,HP-5色谱柱,FID检测器)分析.
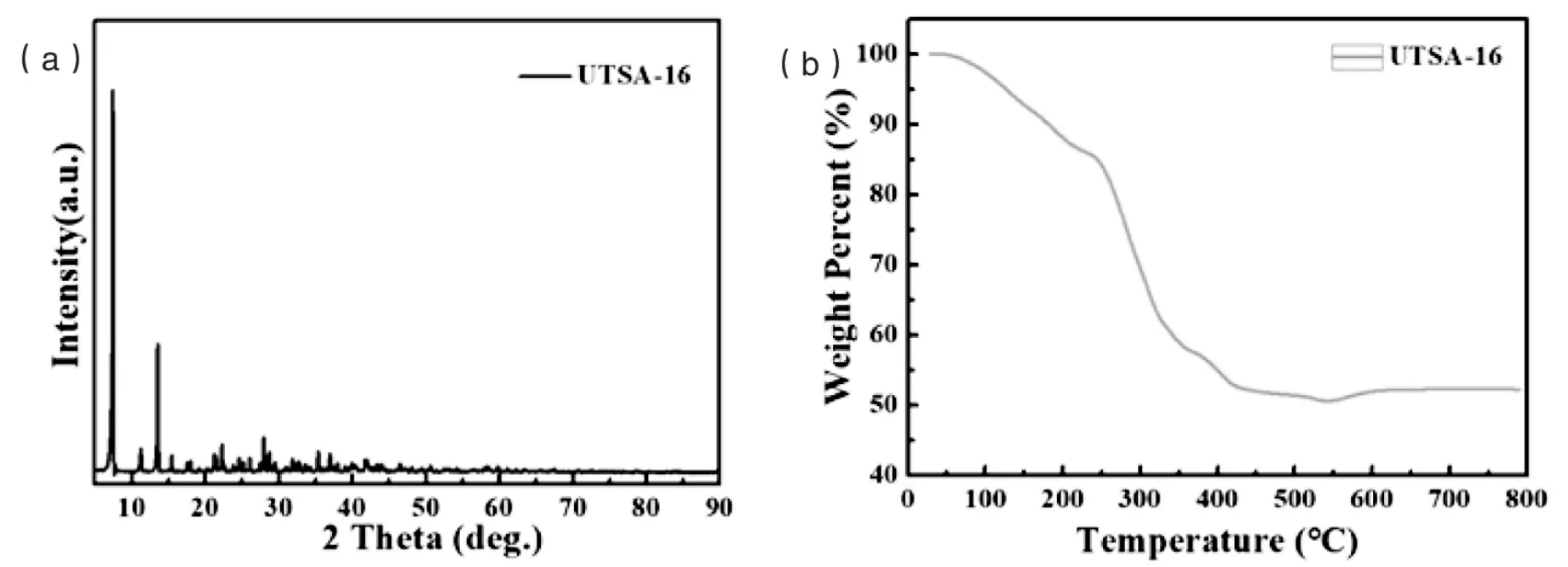
图1 UTSA-16的XRD图谱(a)和热重图谱(b)
2 结果与讨论
2.1 UTSA-16的组成分析
根据文献所报道的方法,我们以柠檬酸和乙酸钴为原料制备了UTSA-16,详细的制备方法和合成过程见实验部分.通过XRD对水热法制备的含钴的金属有机框架材料UTSA-16进行表征测试,结果显示,其中尖锐的衍射峰与文献中所报道的UTSA-16的出峰位置一致(图1a),表明我们成功地制备了UTSA-16,且样品中无其他杂质相,表明样品的结晶性良好[11~12].为了选择合适的UTSA-16的热解程序,我们对样品进行了热重表征测试.如图1b所示,位于100℃附近的失重峰归属于晶体合成的过程中孔道的水分子、乙醇分子的蒸发.在100℃~300℃的温度区间,质量在逐步减少,300℃后快速降低,表明UTSA-16中的有机配体的挥发导致骨架结构坍塌,300℃后质量减少变缓,直到400℃后质量不再减少,表明UTSA-16的完全分解温度为400℃.因此,本文设定UTSA-16的热解程序为:以5℃/min的升温速率,从室温升温到400℃后恒温热解4h.
2.2 Co-P@C的XRD表征
通过程序升温法在400℃下对UTSA-16恒温热解4 h后制备金属前体Co@C,从XRD表征图谱(图2a)可以看出,Co@C 样品在 44.2°、51.5°和 75.9°出现了衍射峰,出峰位置与 Co(JCPDS 15-0806)的(111)、(200)和(220)晶面相对应,表明通过热解UTSA-16成功制备了金属前体Co@C.使用研磨法将次亚磷酸钠与金属前体Co@C研磨均匀混合,再通过程序升温法处理获得Co-P@C纳米催化剂.该催化剂的XRD图谱如图2b所示,Co-P@C样品中含有不同的磷化钴相,主要包括CoP(29-0497)和Co2P(32-0306).在Co-P@C 催化剂的 XRD 图谱(图 2b)中可以看出,在 31.6°、36.3°、48.1°、52.3°、56.0°和 56.8°出现了强的特征衍射峰,其出峰位置与 CoP(JCPDS 29-0497)的(011)、(111)、(211)、(103)、(020)和(301)晶面相对应.同时,在 40.8°、43.3°、44.1°、48.7°、50.4°、52.0°和 56.2°出现了强的特征衍射峰,其出峰位置与 Co2P(JCPDS 32-0306)的(121)、(211)、(130)、(031)、(310)、(002)和(320)晶面相对应,表明通过程序升温法成功地制备了碳包覆的磷化钴纳米催化剂Co-P@C,催化剂中含有CoP和Co2P.

图 2 Co@C(a)和 Co-P@C(b)的 XRD 图谱
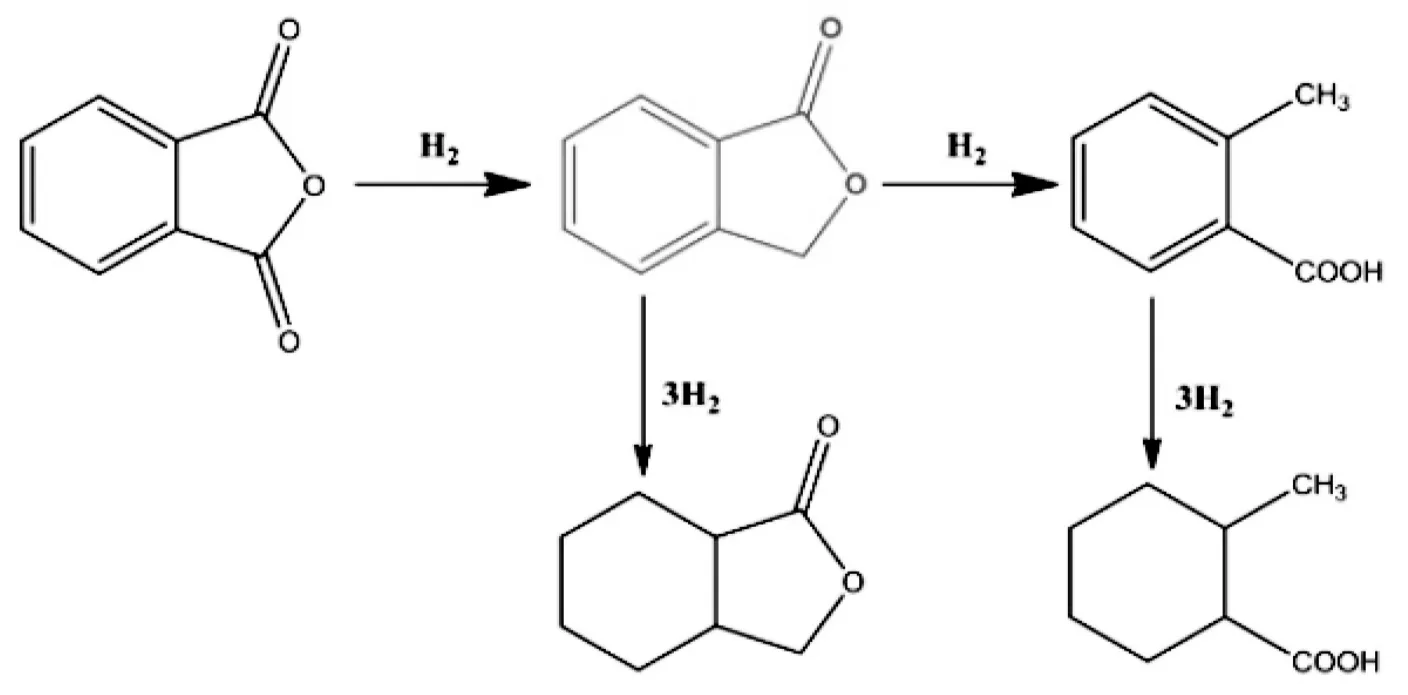
图3 苯酐加氢脱氧的反应网络图
2.3 催化剂活性
将制备的Co-P@C催化剂应用在苯酐的加氢脱氧反应中.苯酐的加氢脱氧反应路径如图3所示,苯酐的加氢脱氧产物主要包括苯酞、邻环己基甲酸甲酯和邻甲基苯甲酸等.
Co-P@C催化剂催化苯酐加氢脱氧的反应结果列于表1之中.在苯酐的加氢脱氧反应中,Co-P@C催化剂在反应温度为220℃,氢气压力为4 MPa和搅拌转速为700 rpm的反应条件下进行催化苯酐加氢反应.随着反应时间的增加,苯酐的转化率逐步提高,催化苯酐加氢脱氧反应5 h后,苯酐的转化率为30.1%,对苯酞的选择性始终保持约42%,邻甲基苯甲酸的选择性为53%,邻甲基苯甲酸是由苯酞的进一步加氢反应生成的,其他产物的选择性小于5%.结果表明,Co-P@C催化苯酐加氢脱氧的反应路径主要为酸酐的加氢脱氧反应,苯环几乎不进行加氢反应.
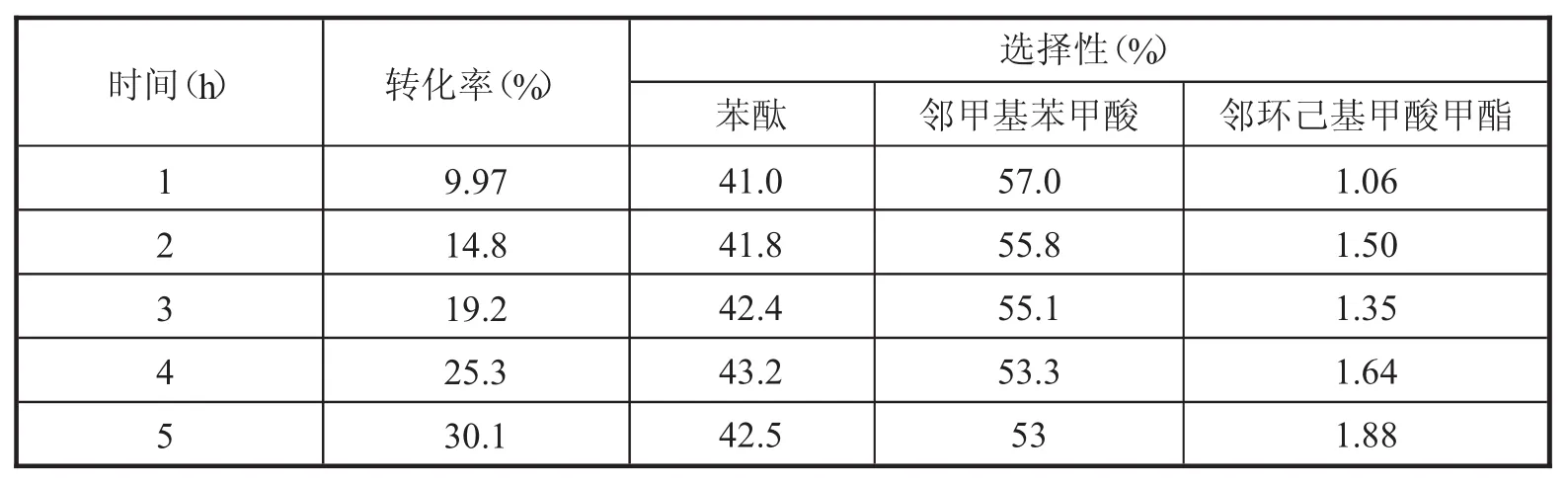
表1 Co-P@C催化苯酐加氢的反应结果
图4显示Co-P@C在220℃下催化苯酐加氢反应随时间变化的产物分布情况.Co-P@C催化苯酐加氢反应5h之后苯酐的转化率为30%.主要产物包括苯酞、邻甲基苯甲酸和邻环己基甲酸甲酯.邻甲基苯甲酸是由苯酞的进一步加氢反应生成的,可进一步发生脱甲基反应形成苯甲酸.

图4 Co-P@C催化苯酐加氢反应中各物质含量随时间变化曲线
3 结论
通过水热法成功地制备了UTSA-16,并以此为金属源,通过程序升温法制备了Co-P@C催化剂并考察其在苯酐加氢脱氧反应中的催化性能,在220℃的反应温度和4.0MPa的氢气压力的优化条件下反应5h后,Co-P@C催化苯酐加氢的转化率为30.1%,苯酞的选择性为42.5%.这表明Co-P@C是一种潜在的加氢脱氧催化剂.
