基于WGCNA 的马铃薯根系抗旱相关共表达模块鉴定和核心基因发掘
2020-07-02秦天元毕真真梁文君李鹏程张俊莲白江平
秦天元 孙 超 毕真真 梁文君 李鹏程 张俊莲白江平,*
1 甘肃省干旱生境作物学重点实验室 / 甘肃省作物遗传改良与栽培种创新重点实验室, 甘肃兰州 730070; 2 甘肃农业大学农学院, 甘肃兰州 730070
外界胁迫(生物胁迫和非生物胁迫)是农业生产面临的主要问题之一, 了解胁迫对植物的影响以及植物对胁迫信号的感知、传递和应答对解决和提高作物产量具有重要的意义。马铃薯作为四大粮食作物之一, 在中国主要种植于年平均降水量小于500 mm 的干旱和半干旱地区, 长期的或季节性的干旱胁迫会严重影响马铃薯的植株长势、块茎产量和商品性[1-2]。马铃薯属浅根系作物, 对干旱胁迫较为敏感, 因此抗旱性研究一直是马铃薯产业关注的问题。研究人员从增大根系、减少蒸腾和提高水肥利用率以提高马铃薯耐旱性开展了大量研究, 也对马铃薯的根系表型和抗逆相关生理生化指标对干旱胁迫的响应情况进行了多种分析, 但针对马铃薯抗旱基因的发掘和响应干旱胁迫的系统性分子机制研究还相对较少, 有待进一步加强[3-4]。随着马铃薯双单倍体参考基因组DM 的构建完成, 利用转录组、蛋白组和代谢组等生物信息学方法研究马铃薯的抗逆性已经成为一个有力的手段, 然而前人的研究大多限于单个调控因子(如顺式表达元件,反式作用因子), 鲜有利用网络分析的方法解析马铃薯抗逆机理的报道。
近年来, 随着高通量测序技术的飞速发展与成本不断降低, 已能采用多样本的转录组测序系统研究生物学问题。传统的2 个样本的比较分析已经不能有效处理海量数据, 因此网络分析应运而生。其中, 权重基因共表达网络分析(weighted gene coexpression network analysis, WGCNA)应用较为广泛,已在多种动物(如恒河猴[5])和植物(草莓[6]、油菜[7]、玉米[8]等)的多样本转录组数据的分析中发挥重要作用。与其他的网络分析不同, WGCNA 可以特异地筛选出与目标性状相关联的基因, 并进行模块化分类,得到具有高度生物学意义的共表达模块, 再从中筛选核心基因。因此, 选择一个可量化的目标性状是WGCNA 分析首要考虑的问题。本研究选用清除活性氧的抗氧化酶活性等生理生化指标作为目标性状。不仅因为抗氧化酶是评价植物耐旱性的传统指标, 而且在近年来模式植物和模式作物的抗旱性研究中, 活性氧(reactive oxygen species, ROS)可作为第二信使, 促使氧化还原信号与其他多种信号通路相互作用, 直接或间接地参与植物的多种代谢过程,共同调节植物对干旱等逆境的响应[9-11]。
本试验以5个时间梯度的干旱胁迫处理, 及根系转录组测序, 结合抗氧化酶活性和根活力的生理生化数据, 利用WGCNA 分析构建共表达基因网络,将基因模块与生理生化数据关联分析, 挖掘出与干旱胁迫调控通路和抗逆指标相关的核心基因, 以期为进一步研究马铃薯根系抗旱的分子遗传机制提供新的线索。
1 材料与方法
1.1 试验材料
由国际马铃薯研究中心引进2 个生育期相同、根系差异明显的马铃薯栽培种C16 (CIP 397077.16)和C119 (CIP 398098.119 (表1), 其组培苗由甘肃农业大学作物遗传改良与栽培种创新重点试验室提供。
1.2 试验方法
1.2.1 试验设计 试验容器是体积250 mL 的罐头瓶, 用150 mmol L-1甘露醇模拟干旱环境。将马铃薯试管苗的茎段接种在正常MS 培养基上, 每瓶接生长状态基本一致的5 株茎段, 为了防止污染,每个处理接5 瓶, 待长至25 d 时, 取3 瓶长势一致的试管苗转入含有150 mmol L-1甘露醇的液体MS培养基中, 处理0 h、2 h、6 h、12 h 和24 h 后, 对根系取样(其中0 h 的样为对照), 样重量不少于2 g,每样3 个生物学重复, 取样后立即置液氮中速冻,-80℃冰箱保存根, 用于RNA 的提取和后续转录组测序, 以及抗逆相关生理生化指标的测定。

表1 试验材料Table 1 Experimental materials
1.2.2 转录组数据的获取与分析 在不同培养条件下, 共采集30 个样品[2 个材料(C16、C119) 5 个时间点(0、2、6、12 和24 h) ×3 次重复]进行转录组测序。以马铃薯基因组及其注释文件作为参考序列,分析转录组测序数据, 所有数据均从Ensembl 网站下 载(http://plants.ensembl.org/Solanum_tuberosum/Info/Index)。首先使用Hisat2 (v2.2.10)将来自干旱处理和对照文库的测序数据和马铃薯参考基因组比对,然后利用 Samtools (v0.1.19)将 SAM 文件转换为BAM 文件, 并对BAM 文件重新排序, 排序后的文件利用cufflinks (v1.2)来组装转录本, 估计这些转录本的丰度, 并且检测样本间的差异表达及可变剪切,再利用python 的一个用于处理高通量数据的HTSeq包来计算样本的表达量。根据基因长度和匹配到该基因的片段数目(readcount 值)计算每一个基因的FPKM (ragments per kilobase of exon per million fragments mapped), 即每1 百万个map 上的reads 中map 到外显子的每1 千个碱基上的reads 的个数。一般情况下, 当基因的FPKM 绝对值大于1 时认为其表达, 否则认为不表达。将表达的基因用DESeq2 R package (v3.5.2)软件和WGCNA 进行不同样本间的差异表达分析、基于抗逆相关生理生化数据的模块划分及核心基因筛选。为了精确定位核心基因, 本试验利用P值小于0.01 的差异表达基因进行后续筛选。采用 AgriGo (http://systemsbiology.cau.edu.cn/agriGOv2/)[12-13]在线软件进行GO 富集分析, 利用Cytoscape[14]软件对基因调控网络进行可视化处理,其中显著富集的GO 类别P值应小于0.05。
2 结果与分析
2.1 差异基因的分析
通过二代转录组测序, 30 个样本中共获得792.64 Gb 和3,801,046,731 个原始测序片段, 对这些原始片段进行过滤后得到Q30 大于95.09%的clean reads 3,720,269,696 个, 它们的GC 含量范围均在41.02%~43.49%之间。30 个样本中, clean reads 与基因组的匹配率皆大于85%, 说明所有试验样品的采集及测序结果高度可信。为了筛选不同处理相比于对照和不同品种间的差异表达基因, 基于每个转录本的readcount 值, 利用DESeq2 软件进行目的样本间的差异表达基因分析, 以校正后的P值小于0.01为阈值, 以差异倍数|log2FC| > 1 为条件, 筛选出差异极显著的转录本用于后续分析。
将C16 和C119 在不同干旱处理时间点的数据分别与未处理的数据比对, 筛选出极显著上调差异表达(图 1-A, B)和极显著下调差异表达的基因(图1-C, D)。干旱处理2 h 时, C16 中上调差异表达基因为97 个, 远远少于C119 中的421 个(图1-A,B)。在持续胁迫到24 h 时, 上调差异表达基因数达到最大值, C16 中为606 个, 而C119 中为843 个,在处理2 h、6 h、12 h 和24 h 四个时间点, 相对于未处理都上调的基因在C16 中有49 个, 而C119有78 个, 说明在短期干旱处理后C16 根系中上调表达基因数目远少于C119。而下调差异表达的基因, 胁迫2 h 时 C16 中为12 个, 少于C119 中的50 个(图1-C, D)。在持续胁迫到24 h 时, C16 中下调表达的差异基因为1315 个, 而C119 中为1718个, C16 在持续处理2 h、6 h、12 h 和24 h 四个时间点相对于0 h 都下调的基因为42 个, 而C119 为21 个, 对比发现, 在短期处理后C16 根系中上下调表达基因数目远少于 C119。以上结果说明, 相比于C16, C119 在甘露醇模拟的干旱胁迫下有更多的基因积极上下调表达, 以调整体内代谢途径的变化来防御逆境伤害。
2.2 差异基因的WGCNA 分析
WGCNA 分析, 即权重基因共表达网络分析,是一种多个样本基因表达模式的分析方法。通过WGCNA 分析可将表达模式相似的基因聚类而形成不同的模块, 分析模块与特定性状或表型之间的关联性, 通过基因聚类和关联分析结果构建共表达调控网络。相比于只关注差异表达基因的传统分析方法, WGCNA 利用数千或近万个变化最大的基因或全部基因分类为不同的基因集, 并与表型进行关联分析, 既充分利用了整体组学信息, 也把数千个基因与表型的关联转换为数个基因集与表型的关联,免去了多重假设检验校正。位于调控网络中心的基因被称为核心基因, 即hub gene, 这类基因通常是关键的调控基因, 是值得我们深入挖掘和分析的对象, WGCNA 分析在组学研究中被广泛应用。本研究将C119 和C16 的根系中响应干旱胁迫的差异基因结合其根系抗逆生理生化指标数据(附表1), 通过WGCNA 分析和基因集-表型关联分析, 深入挖掘出马铃薯根系中抗旱调控网络的核心基因。
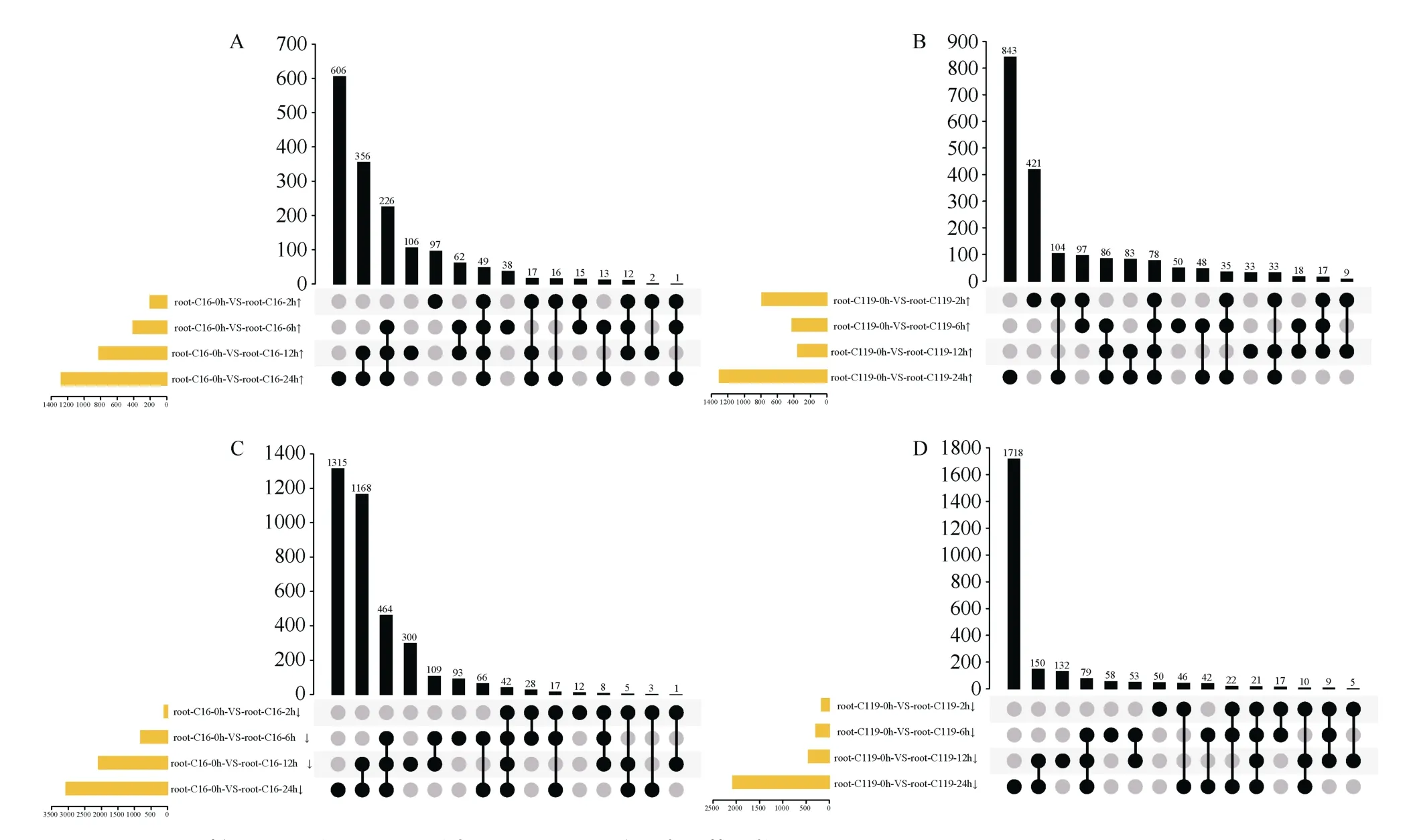
图1 不同处理时间下C16 与C119 根系中上调和下调的差异表达基因数Fig. 1 Upregulated and down-regulated genes in roots of C16 and C119 under different treatments
2.2.1 去除离群样本 由于网络模块分析结果容易受到离群样本的影响, 因此在构建网络模块之前须去除离群样本, 以保证结果的准确性。本研究共有30 个样本, 通过计算各个样本表达水平的相关系数后聚类(图2-A)。相关性较低的样本, 或者在树形图上无法聚类的样本即为离群样本, 如root.C119.6h.rep1、root.C119.12h.rep3、root.C16. 2h.rep1、root.C16.6h.rep3、root.C16.0h.rep1、root.C16. 12h.rep3。离群样本被去除后, 剩余样本的聚类树如图 2-B所示。
2.2.2 基因共表达网络中软阈值的确定 选取C16 和C119 两个品种的根系在未处理和短期干旱处理2、6、12 和24 h 得到的基因表达谱矩阵来构建差异基因的共表达网络, 需要计算出每个基因两两之间的相关系数, 进而采用公式Smn= cor(xm,xn),S=[Smn]得到基因之间的相似矩阵, 式中Smn表示基因m和基因n 之间的皮尔森相关系数,S表示相似矩阵。利用R 软件中的WGCNA 包来构建权重基因共表达网络。为了使网络符合无尺度网络分布, 利用WGCNA 包中的函数pick Soft Threshold 来计算权重值。根据图3 的结果, 选择软阈值β = 9 来构建共表达网络。
2.2.3 基因共表达网络基因聚类和模块切割 确定软阈值β=9 之后, 根据公式为Amn= [(1+Smn)/2]β将相似矩阵转换为邻接矩阵, 再将邻接矩阵转为拓扑重叠矩阵(Topological Overlap Matrix, TOM),同时利用函数diss Tom = 1-TOM 对拓扑重叠矩阵取逆得到相异性矩阵, 这样做的目的是为了清除背景噪音和伪关联带来的误差。最后利用函数hclust 对相异矩阵进行层次聚类, 采用动态切割法(Dynamic Tree Cut)将产生的聚类树切割, 该过程可以把表达模式相近的基因合并在同一个分支上,每一个分支代表一个共表达模块, 不同的颜色代表不同的模块。差异基因根据其FPKM 值进行相关度分析并聚类, 相关度较高的基因被分配到同一个模块中(图4)。

图2 样本的聚类树Fig. 2 Dendrogram of samples

图3 基因共表达网络软阈值的确定Fig. 3 Soft threshold determination of gene co-expression network
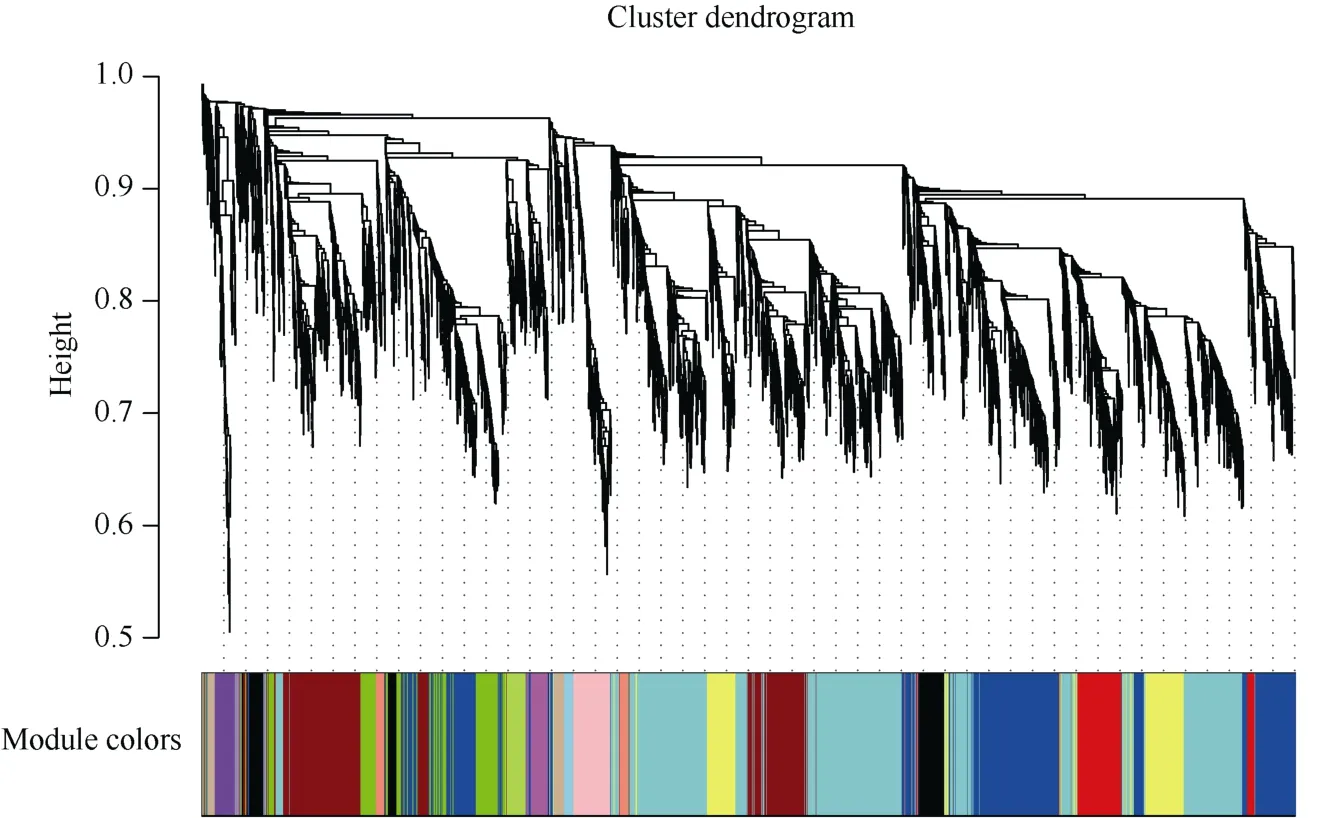
图4 基因共表达网络基因聚类数和模块切割Fig. 4 Gene co-expression network gene clustering number and modular cutting
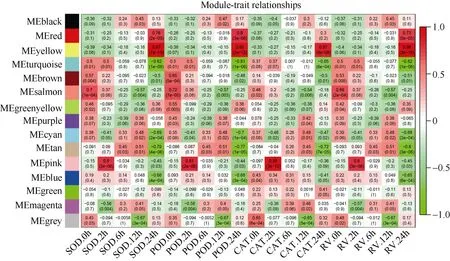
图5 基因共表达网络模块与生理生化性状的关联热图Fig. 5 Association analysis of gene co-expression network modules with physiological and biochemical traits
2.2.4 基因共表达网络中模块与生理生化性状矩阵的关联分析 将上一步所获模块中按照75%的相关性兼并融合, 结合样本的4 个生理生化性状[超氧化物歧化酶(superoxide dismutase, SOD)、过氧化物酶(peroxidase, POD)、过氧化氢酶(catalase, CAT)和根活力(root vigor, RV)]在不同处理时间下的矩阵得到15 个与这些性状相关联的模块, 不同的模块分别以不同的颜色表示(图5)。其中, 部分模块与生理生化性状高度关联。例如, Yellow 模块内的基因表达模式随着处理时间的增加与生理生化性状的相关度存在由负到正的趋势, 并在24 h 时与SOD、POD、CAT和RV 均呈极显著正相关(分别为r=0.97,P=7E-15;r=0.95,P=2E-12;r=0.87,P=4E-08 和r=0.98,P=3E-16), Red 模块与与生理生化性状的关联情况与Yellow 模块类似; Turquoise 模块内的基因表达模式则随着处理时间的增加与生理生化性状的相关度存在由正到负的趋势, 并在与处理24 h 时与SOD、POD、CAT 和根活力(RV)均呈极显著负相关(分别为r=-0.82,P=1E-06;r=-0.81,P=1E-06;r=-0.65,P=6E-04 和r= -0.82,P=1E-06); Blue 模块与生理生化性状的关联情况与Turquoise 模块类似; Pink 模块与处理2 h 时的SOD、POD、CAT 和根活力(RV)存在高度正相关(分别为r=0.80,P=2E-06;r=0.81,P=2E-06;r=0.95,P=3E-12 和r=0.80,P=3E-06)(图5)。
2.2.5 通过特征向量基因分析确定目标基因模块
基因模块是指一系列高度相关基因的集合, 同一个模块中的基因可能具有相似的生物学功能。由于具有相似表达模式的基因可能具有相似的生物学功能, 通过WGCNA 构建的模块在很大程度上与特定的性状相联系, 而特定的性状又与特定的生命活动存在高度协同性。因此, 为了进一步解析生理生化性状特异性模块的生物学功能, 根据基因共表达网络模块与时间性状的关联热图, 挑选出与性状关联度较高的模块, 并进行下一步研究。
由于模块中的基因数目庞大, 可利用降维的思路来研究基因模块, 即把模块中具有代表性的基因—特征向量基因(module eigengene, ME)挑选出来。用ME 代表模块中成千上万的基因进行相关性分析,利用这种方法可以进一步理清基因模块与模块之间的相互关系及与不同处理时间的表型性状的相关性, 筛选出目标基因模块。对所有模块的ME 进行聚类分析后发现, 部分ME 之间存在很高的相关性(图6)。ME 之间的相关性越高, 其所在的模块的相关性也就越高, 通过两两之间的ME 相关性分析,发现Red 模块与Blue 模块中ME 之间的相关性达到0.87, Yellow 模块与Turquoise 模块中ME 之间的相关性达到0.88 (图7), 且Red、Yellow 模块内基因随着干旱处理时间的增加, 基因表达模式与抗逆相关生理生化性状由负相关逐渐转为极显著正相关,Tuqurise、Blue 模块内基因随着处理时间的增加, 基因表达模式与目标性状由正相关逐渐转为极显著负相关, 因此这4 个模块可作为目标基因模块。
将这4 个模块中的所有基因和ME 基因在所有样本中分别进行了表达水平分析, 发现每个模块内的基因表达水平高度相关, ME 表达水平与模块整体表达水平也高度相关, 说明目标模块的ME 可充分代表其模块中的整体基因(图8)。
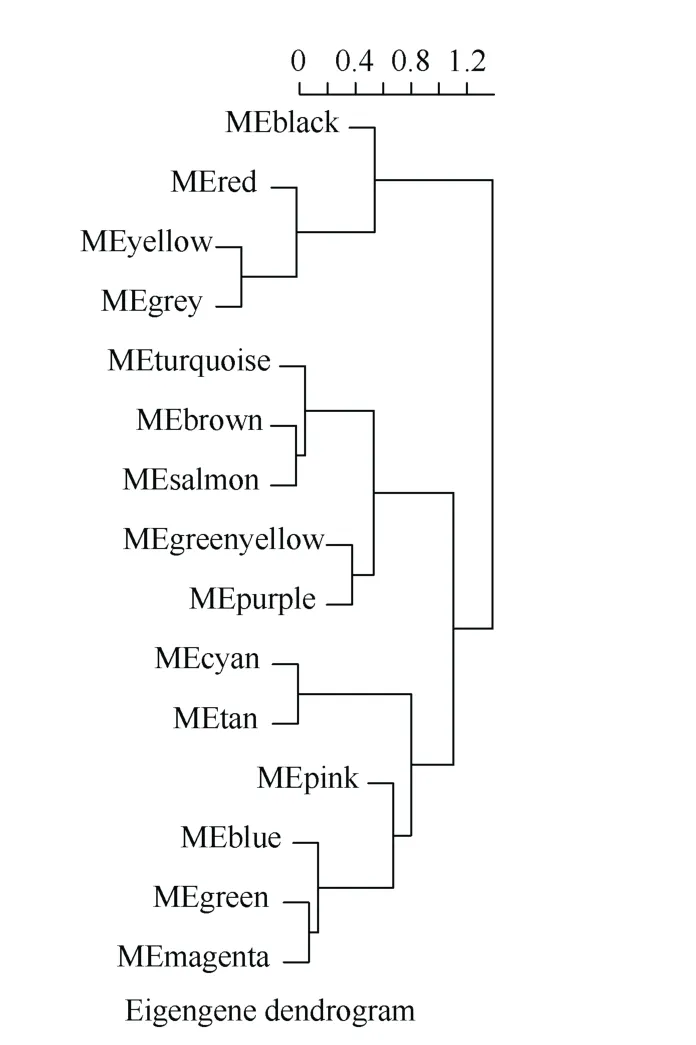
图6 ME 聚类树Fig. 6 ME cluster tree
2.2.6 目标基因模块的GO 注释 为进一步挖掘目标基因模块的功能, 利用AgriGo 在线工具对筛选出的4 个目标基因模块Red、Yellow、Turquoise 和Blue 进行GO 功能富集分析。结果表明这4 个模块都可以显著富集到生物学过程、分子功能以及细胞组分三大分类下的若干GO 通路(图9)。如表2 所示,Red 模块可以富集到氧化还原相关基因(GO:0016705)和内肽酶抑制剂活性基因(GO:0004866)等调控通路; Yellow 模块可以富集到与蛋白质结合相关的通路(GO:0005515); Turquoise 模块可以富集到光合作用(GO:0015979), 也富集到与氧化还原酶活性相关的通路(GO:0016705), 还富集到类固醇合成途径中催化类固醇脱氢反应(以类固醇羟基为氢供体, 以NAD 或NADP 为氢受体)的脱氢酶及相关通路(GO:0006694、GO:0003854、GO: 0033764、GO:0016229、GO: 0016614、GO: 0016616 和GO:0051287);Blue 模块可以富集到与氧化还原酶活性相关的基因(GO:0016705) 和 与 细 胞 骨 架 相 关 的 基 因(GO:0007010 和GO:0015629)。总体来看, 4 个模块都富集到了氧化还原酶活性及细胞骨架组织相关的通路, 说明WGCNA 可以有效构建到具有生物学意义的共表达模块, 这些模块可成为本研究的重点关注对象。
2.2.7 目标模块中核心基因的筛选和功能分析
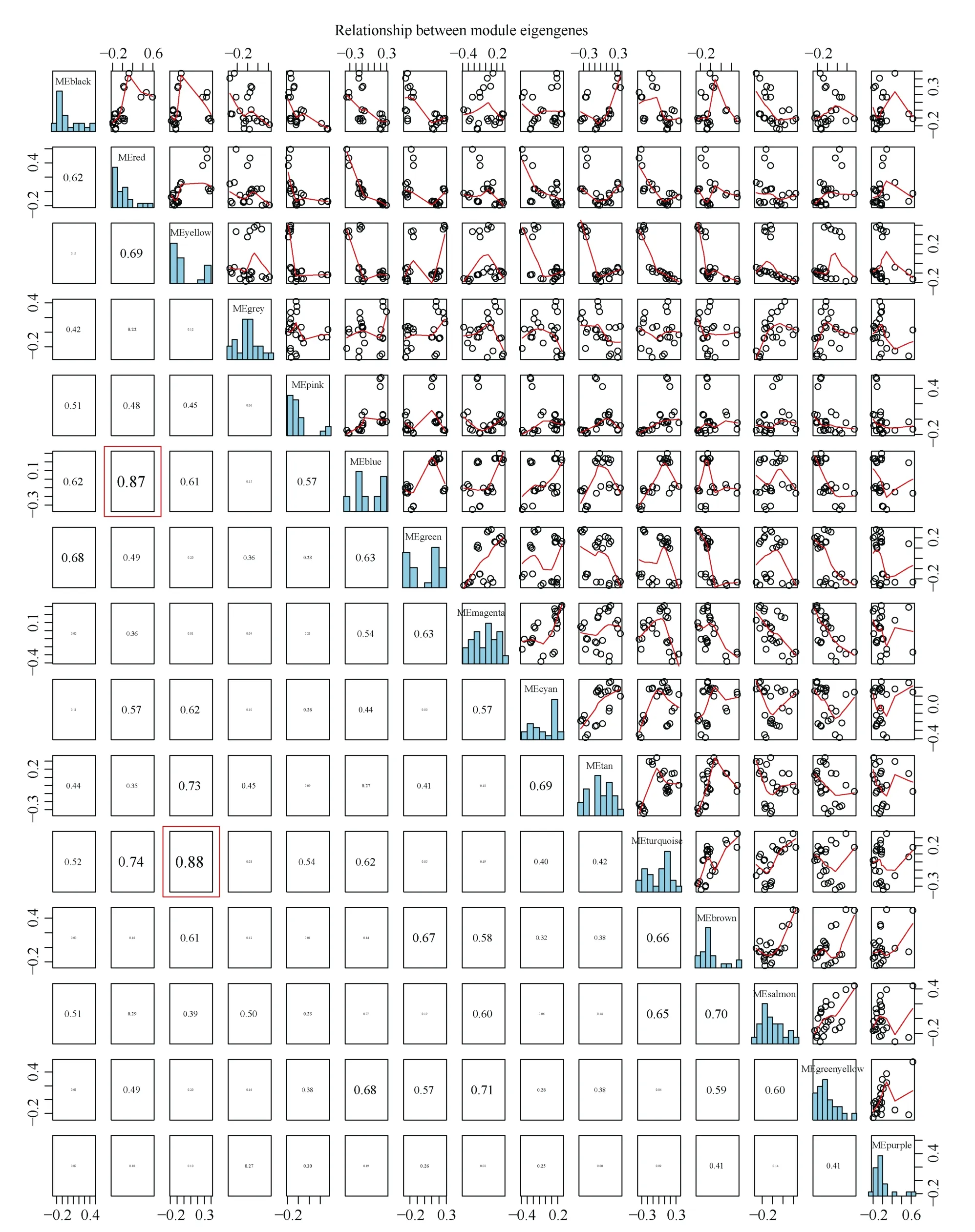
图7 不同模块两两之间ME 的相关性Fig. 7 ME correlation between different modules
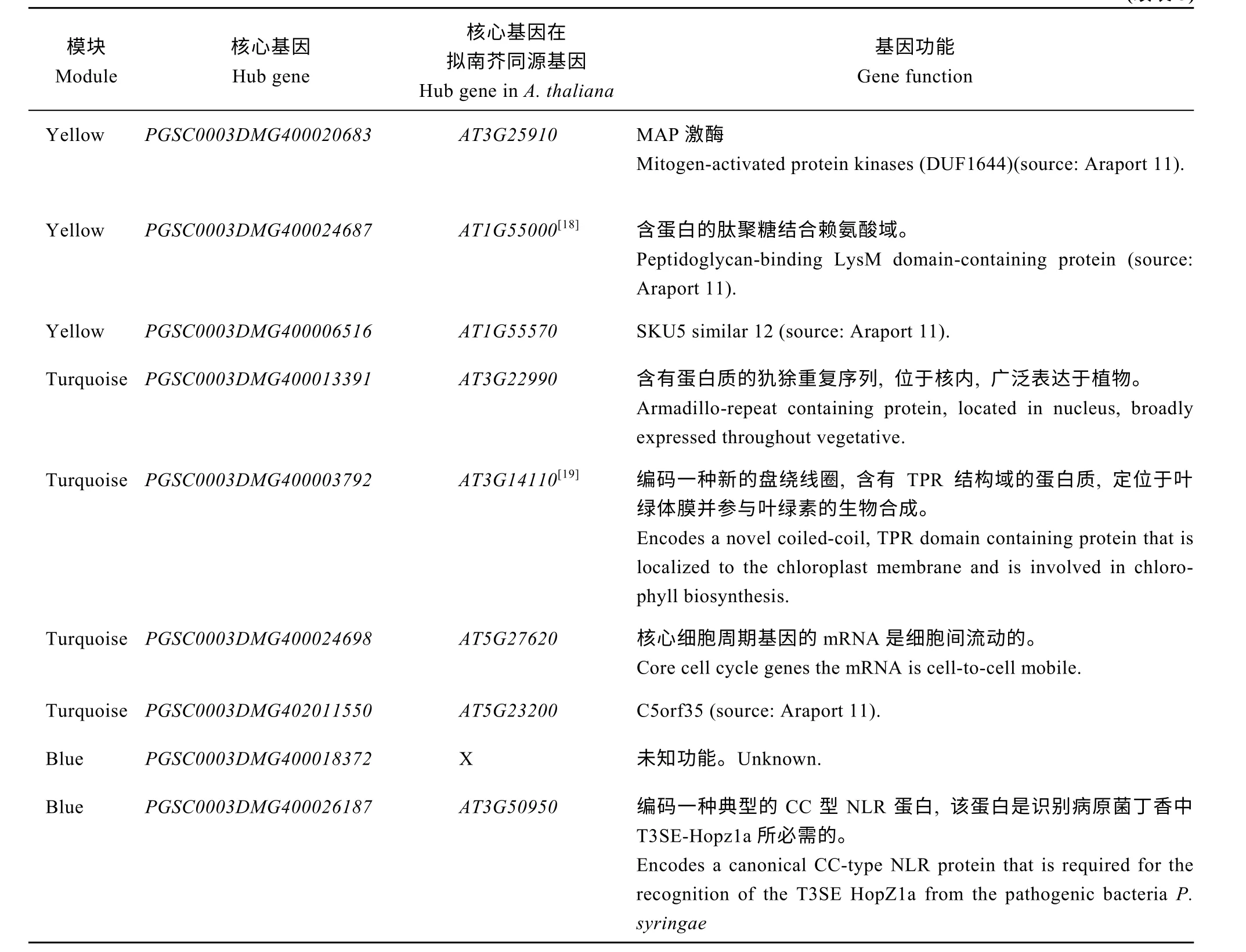
为了获得上述 4 个模块中的核心基因, 利用Cytoscape 软件对基因调控网络进行可视化处理, 筛选出模块中高连通性的基因, 这些基因则作为该模块中的核心基因(图10 和图11)。在这些网络中, 每一个节点代表一个基因, 节点通过线相联系, 处于连线两端的基因通常被认为具有相同的生物功能。其中在Red 模块中筛选出了5 个核心基因, Yellow模块中筛选出了9 个核心基因, Turquoise 模块中筛选出4 个核心基因, Blue 模块中筛选出了2 个核心基因,为了获取这些核心基因的功能信息, 我们利用马铃薯基因数据库(http://solanaceae.plantbiology.msu.edu/index.shtml)和 NCBI 数据库(https://www.ncbi.nlm.nih.gov/)查询这些核心基因的相关报道, 并借助TAIR 数据库(https://www.arabidopsis.org/)注释这些核心基因在拟南芥中的同源基因的功能(表3), 通过已报道的文献, 本研究发现这些筛选出的核心基因基本都与干旱胁迫相关, 其中PGSC0003 DMG400026572在拟南芥中的同源基因AT3G51730基因参与了植物液泡的形成, 而液泡在植物生长、发育和应激反应中起着中心作用。PGSC0003DMG 400029350在拟南芥中的同源基因AT1G05680基因通过调节活性氧和氧化还原信号与植物激素发生协同或拮抗作用, 引起植物对生物和非生物胁迫的保护反应。PGSC0003DMG400006704在拟南芥中的同源基因AT5G35200基因通过诱导启动蛋白磷酸化调节下游的信号包括穿过质膜的离子通量、MAPK 的活化和活性氧的形成。PGSC0003 DMG400024687在拟南芥中的同源基因AT1G55000基因与泛素化的特异性相关, 能促进泛素转移到合适的靶点进而对细胞活动进行调控。PGSC0003 DMG400003792在拟南芥中的同源基因AT3G14110基因与植物体内的活性氧的变化相关, 在逆境环境下, 该基因可激活多种应激反应。
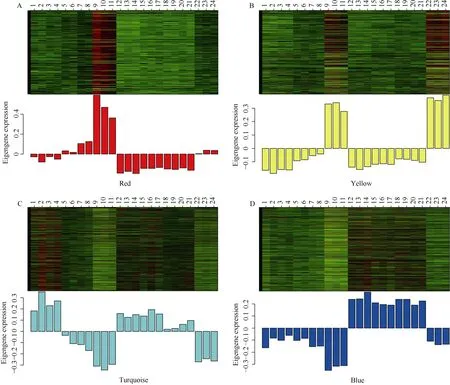
图8 各样本中不同模块的所有基因与相应ME 的表达水平Fig. 8 Expression levels of all genes and corresponding ME in different modules of each sample

图9 目标模块内基因GO 注释Fig. 9 Gene GO annotation in target module

表2 目标模块的部分GO 富集分析结果Table 2 Partial GO enrichment analysis of target modules

(续表2)

图10 Red 和Yellow 模块内的基因共表达网络及其核心基因Fig. 10 Gene co-expression network and hub genes in Red and Yellow modules

图11 Turquoise 和Blue 模块内的基因共表达网络及其核心基因Fig. 11 Gene co-expression network and hub genes in Turquoise and Blue modules

表3 不同模块中核心基因的功能注释Table 3 Functional annotations of hub genes in different modules
2.2.8 核心基因的RT-qPCR 验证 从相关性最高的4 个基因模块中筛选到20 个可能与干旱胁迫相关的核心基因(表3), 从中挑选了14 个基因进行实时荧光定量PCR 验证。将用于转录组测序的备份RNA 反转录, 通过q-PCR 检测这些基因在C16 和C119 中对干旱胁迫的响应情况。最终结果的变化趋势与转录组结果基本一致(表4 和图12), 进一步证明了转录组测序结果的可靠性, 并验证了这些核心基因确实响应干旱胁迫。
(续表3)

表4 字母与基因的对应表Table 4 Correspondence between letters and genes
3 讨论
干旱胁迫是影响自然界植物地理分布, 限制农业生产力和威胁粮食安全的主要环境因素之一。全球气候变化更加剧了干旱胁迫的不利影响, 预计还将导致极端天气的频率增加[20-21]。马铃薯作为第四大粮食作物, 在我国的主产区也处在气候变化的高风险区域, 干旱胁迫是主要的风险之一[22]。因此, 提高马铃薯的抗旱性, 解析其在干旱胁迫下的感知和适应机制对提高农业生产力和节约环境资源均具有重要意义。
植物根系是植物汲取和转化水分和营养的主要器官, 也是植物面临土壤胁迫环境时首先感知胁迫信号的器官, 在植物应对非生物逆境尤其是干旱胁迫时起关键作用[23-25]。虽然马铃薯根系抗逆遗传机理的解析取得了一定的进展, 例如通过数量性状位点定位和关联分析等方法鉴定了许多重要的根系抗逆基因[26-27], 但是这些研究手段都有一定的局限性,例如数量性状定位不能反映遗传背景复杂群体的遗传异质性, 此外, 大多数的农艺性状还与整个群体结构存在很高的相关性, 而利用现在生物信息技术和数据分析方法则可以通过共表达模块化处理, 将成千上万具有相似生物学功能的基因分配到同一个模块中, 通过研究模块的生物学意义来进一步挖掘模块内基因的功能, 较好地解决了对遗传背景复杂群体的性状解析不足的问题, 因此被广泛地应用于大数据组学的研究之中[28-30]。因此, 本研究干旱处理马铃薯幼苗后, 收集其根系材料进行转录组分析,以期获得马铃薯根系抗逆相关的新的研究线索。与传统的仅对比处理前后的转录组差异分析不同, 本研究针对2 个品种, 分别设置了4 个处理时间梯度,加上未处理的对照, 每样3 个生物学重复, 共计30个样本进行了转录组测序。与此同时, 本研究还检测了所有处理时间下每个样本的胁迫相关生理生化指标(SOD、POD、CAT 和RV)。
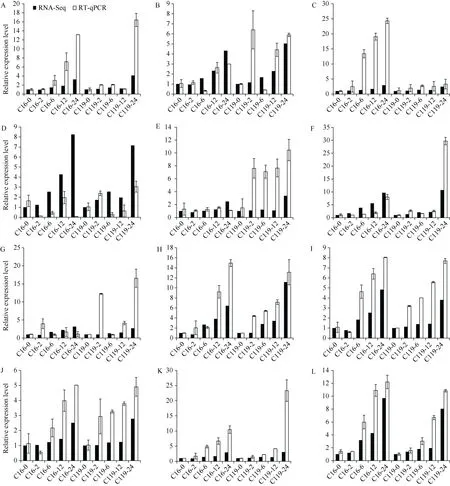
图12 核心基因的RT-qPCR 验证Fig. 12 RT-qPCR validation of Hub genes
大量的转录组数据和生理数据获得后, 如何从中挖掘蕴含的生物学意义成为首要考虑的问题。网络分析在基因组、转录组和代谢组等高通量组学数据的发掘中应用较为广泛。为了发掘与抗旱相关的基因或调控通路, 本研究采用了WGCNA 分析方法,可特异地筛选出与目标性状相关的基因, 并进行模块化分类, 得到具有高度生物学意义的共表达模块,已经被证明是一种高效的数据挖掘手段[31]。ROS 的产生是植物受到干旱胁迫后最早的反应之一, ROS是许多胁迫相关生物合成、信号转导和代谢相关途径的重要调节剂, 其过量积累对细胞具有毒害作用[32], 因此维持其动态平衡是植物应对胁迫的重要调节方式, 而SOD、POD 和CAT 作为清除ROS的抗氧化酶, 可较为直接地反映植物对胁迫的响应情况[33], 可作为WGCNA 分析中的目标性状。因此,我们利用WGCNA 分析方法, 富集到了15 个与生理性状相关联的共表达模块, 其中4 个模块与这些抗逆性状高度相关, 通过进一步的网络分析, 找到了这4 个模块的核心基因。表明WGCNA 可以构建到具有生物学意义的共表达模块和通路, 为下一步深入研究打下基础。
本研究通过WGCNA 富集到的15 个与胁迫相关生理生化指标(SOD、POD、CAT 和RV)相关联的共表达模块, 由于模块较多, 因此挑选了4 个与生理性状关联度最高的模块进行下一步研究。模块内的基因表达模式随着处理时间的增加, 存在与生理性状由负相关逐渐转为极显著正相关的Yellow 模块和Red 模块, 以及存在相反趋势的Turquoise 模块和Blue 模块。由于每个模块的基因数量庞大, 本研究利用各个模块的特征向量基因对模块两两之间进行相关性分析, 结果表明, 这4 个模块间的确存在极显著的相关性且关联度最高, 进一步印证这4 个模块可作为根系抗逆性状机理研究的目标模块。通过对这4 个模块的GO 功能富集分析发现, 均可以得到与抗逆相关的具有生物学意义的调控途径。例如,Red 模块、Turquoise 模块和Blue 模块都富集到与氧化还原活性相关的通路(GO:0016705), Turquoise 模块还富集到类固醇合成途径中催化类固醇脱氢反应的脱氢酶及相关通路(GO:0006694)。有研究表明, 在拟南芥和玉米中过量表达油菜素固醇合成途径的脱氢酶基因, 可导致ABA 的积累, 增强植物抗性, 提高玉米在逆境下的产量[34-35]。说明WGCNA 分析的确可富集到与目标性状相关的具有生物学意义的共表达模块。
本研究针对4 个与目标性状关联度最高的模块分析表明, 其核心基因包含干旱胁迫相关基因, 例如Red 模块中的参与木质部分化的基因(PGSC0003 DMG400020562)、Yellow 模块中的 MAP 激酶(PGSC0003DMG400020683)、Turquoise 模块可能参与叶绿素合成的基因(PGSC0003DMG400003792)。其中Yellow 模块中PGSC0003DMG400029350基因在拟南芥中的同源基因AT1G05680可编码一种UDP-葡萄糖基转移酶UGT74E2, 在拟南芥中的研究表明, 其可被过氧化氢或非生物胁迫诱导表达,通过对生长素吲哚-3-丁酸(IBA)和吲哚-3-乙酸(IAA)的动态平衡调节, 参与氧化还原信号与植物激素信号的整合互作, 增加植物在非生物逆境中的保护反应[36]。这些基因在模式植物拟南芥和其他作物如玉米、水稻等中有较多的研究, 在马铃薯中的报道还相对较少, 可进行深入探究。
以上结果表明, 利用WGCNA 分析, 可挖掘到与目标性状的生物学意义高度关联的基因模块和核心基因, 为挖掘目标基因提供新的研究思路, 为解析复杂的农艺性状提供参考。在本研究中, 重点关注了Red、Yellow、Turquoise 和Blue 四个相关性高的调控模块, 其余的基因模块虽然没有被详细论述,但也可能包含与根系抗逆相关的通路, 可进一步挖掘其蕴含的生物学意义。
4 结论
本研究构建了与抗逆生理性状相关联的权重基因共表达网络, 得到了15 个与根系抗旱密切相关的基因模块并进行了GO 通路富集。挑选出4 个与目标性状关联度最佳的基因模块进行深入分析, 其中在Red 模块中挑选出了5 个核心基因, Yellow 模块中挑选出了9 个核心基因, Turquoise 模块中挑选出4个核心基因, Blue 模块中挑选出了2 个核心基因, 通过功能注释, 发现部分核心基因与已报道非生物胁迫调控通路紧密相关, 还有一部分功能仅在模式植物中有一些研究。本研究结果可为马铃薯根系抗旱的分子机制研究提供线索, 也为耐旱性马铃薯新品种培育提供理论支持。
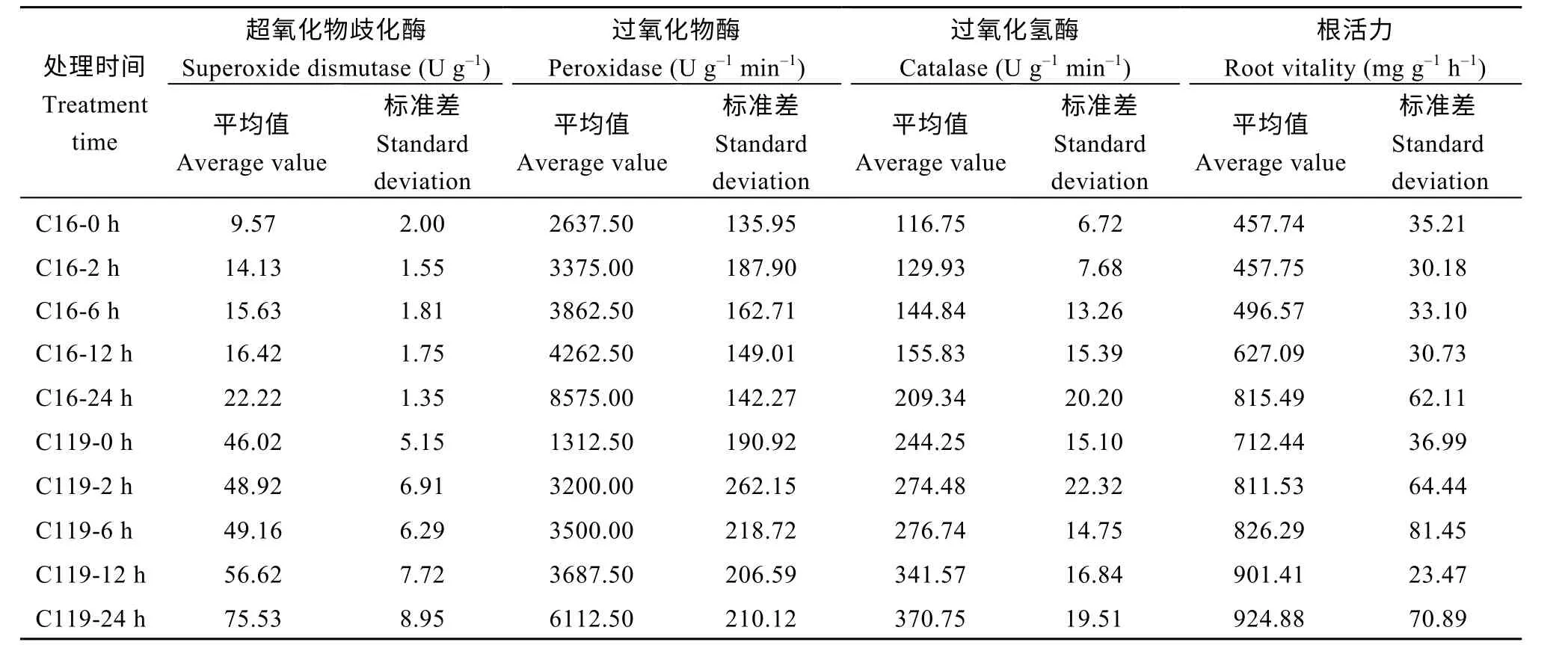
附表 1 C16 和C119 在不同处理时间下的生化指标数据Supplementary table 1 Biochemical indicator data for C16 and C119 at different treatment times
