类似卟啉病的肝豆状核变性1例报告
2020-05-29王英灏陈若蝉肖满意汪玲
王英灏,陈若蝉,肖满意,汪玲
(1.中南大学湘雅二医院 眼科,湖南 长沙 410011;2.中南大学湘雅医院 感染病科,湖南 长沙 410008)
肝豆状核变性(hepatolenticular degeneration,HLD)又名威尔逊病(Wilson's disease,WD),是一种常染色体隐性遗传的铜代谢障碍疾病。其发病机制是由于位于第13 号染色体的编码铜跨膜转运蛋白-ATP7B基因突变,从而导致体内铜离子转运及排泄障碍,导致其在肝脏、神经系统以及其他组织器官中过度沉积,从而引起各种各样的临床表现,缺乏特异性。卟啉病(porphyria)又名紫质病,是一种常染色体显性遗传的代谢紊乱疾病。它是由于血红素生物合成途径中的酶活性缺乏,引起卟啉或其前体浓度异常升高,并在组织中蓄积并引起损伤的一组疾病。其临床表现同样罕见,多变,缺乏特异性,早期诊断困难,易误诊和漏诊。两种疾病均被列入国家卫生健康委员会制订的《罕见病诊疗指南(2019 年版)》[1-2],现将本院近期收治的1 例临床表现似卟啉病的成人肝豆状核变性病例报告如下。
1 病例资料
患者雷XX,18 岁;女,因颜面部、双手麻木9 d,腹痛伴皮肤巩膜黄染5 d 于2019 年9 月14日入住本院。患者于2019 年8 月30 日于室外活动后出现额部、面颈部及双手掌的麻木感,颜面日晒部位出现明显灼烧感、瘙痒,以及小水疱,自行按皮肤晒伤处理后好转。9 月9 日晚患者突发剧烈腹痛,烦躁不安,就诊诊所排除急腹症,数小时后腹痛自行缓解,之后出现尿色深黄。9 月12 日再次出现剧烈腹痛数小时,家人发现其皮肤巩膜黄染,遂送往本院就诊。既往史:患者孕期常规孕检多次发现转氨酶升高,在外院完善相关检查,考虑肝内胆汁淤积症(intrahepatic cholestasis of pregnancy,ICP),间断口服熊去氧胆酸治疗,自觉无明显作用自行停用。2019 年7 月底患者因腹痛考虑ICP 于连续硬脊膜外麻醉下行剖宫产术,产下1 男婴,体重2 700 g,阿普加评分良好。患者为初中学历,学习成绩一般,否认肝炎病史,家族遗传病史不详,父母非近亲结婚。
入院时体查:体温:36.5℃;脉搏:80 次/min;呼吸:17 次/min;血压:123/74 mmHg(1 mmHg=0.133 kPa);体重:66 kg,发育正常,神志清楚,查体合作,贫血貌,定向力正常,计算力下降,未引出扑翼样震颤,可见肝掌,头皮和前额处可见少量散在丘疹,留有色素沉着,皮肤巩膜深度黄染。心肺无异常,腹部稍隆起,腹肌柔软,右上腹有轻压痛,墨菲氏征阴性,全腹无反跳痛。肝脏肋下3 cm,质地中等,无触痛,脾脏肋下刚可触及,质软,边界清楚,无触痛,移动性浊音阳性,肠鸣音正常。双侧下肢轻度水肿,脊柱、四肢、肛门外生殖系统检查未发现异常。
完善相关检查:血常规:红细胞2.55×1012/L,血红蛋白89 g/L,血小板174×1012/L;肝功能:谷丙转氨酶2.5 u/L,谷草转氨酶61.7 u/L,总蛋白58.5 g/L,白蛋白32.9 g/L,总胆红素464 μmol/L,直接胆红素255.8 μmol/L,总胆汁酸178 μmol/L;凝血功能:凝血酶原时间23.7 s,凝血酶原活动度34.44%,国际标准化比值1.91;CA199 307 u/mL;贫血四项:铁蛋白452.3 ng/mL;叶酸5.15 ng/mL;促红细胞生成素113.61 mIU/mL;维生素B12>1 500 pg/mL;风湿免疫全套:补体C3、C4 下降,C-反应蛋白及降钙素原轻度升高,24 h 尿铜2 921.5µg,血清铜蓝蛋白66 mg/L。大便常规、尿常规、血氨、血沉、输血前四项、病毒性肝炎病原学、EB 病毒、血小板抗体、自免肝全套、ANA 谱及狼疮全套、Ham+Coomb's 试验、PNH 克隆流式检测正常。尿液送皮肤科Wood 灯照射显示无明显改变。眼部裂隙灯检查发现Kayser-Fleischer(K-F)环阳性。腹部彩超:右肝大、肝实质弥漫性病变;左肝侧枝循环;脾大、腹水;胆囊沉积物,胆囊炎;可排除“布加”氏征。胸腹部+盆腔增强CT:①胆囊炎:胆囊窝积液;②肝脏密度较均匀减低,格里森鞘增宽:活动性肝炎所致?③脾大;④腹、盆腔积液。腹部MRI:①肝实质弥漫性小结节状改变,考虑肝硬化?肝实质损害?②胆囊胆固醇结晶沉积;③腹膜后多发稍大淋巴结。遗传病基因检测报告中,患者遗传病基因测序报告示:杂合突变ATP7Bc.51+2T>G,高通量检测结果提示受检者chr13q14.3(52158814-53624916)区域内可能存在片段的杂合缺失,长度约1.47 Mb,包含ATP7B基因。之后召集患者家属进行基因测序,报告提示患者父亲及妹妹杂合突变ATP7Bc.51+2T>G;患者儿子chr13q14.3(52158814-53624916)区域内可能存在片段的杂合缺失,长度约1.47 Mb,包含ATP7B基因,见图1。
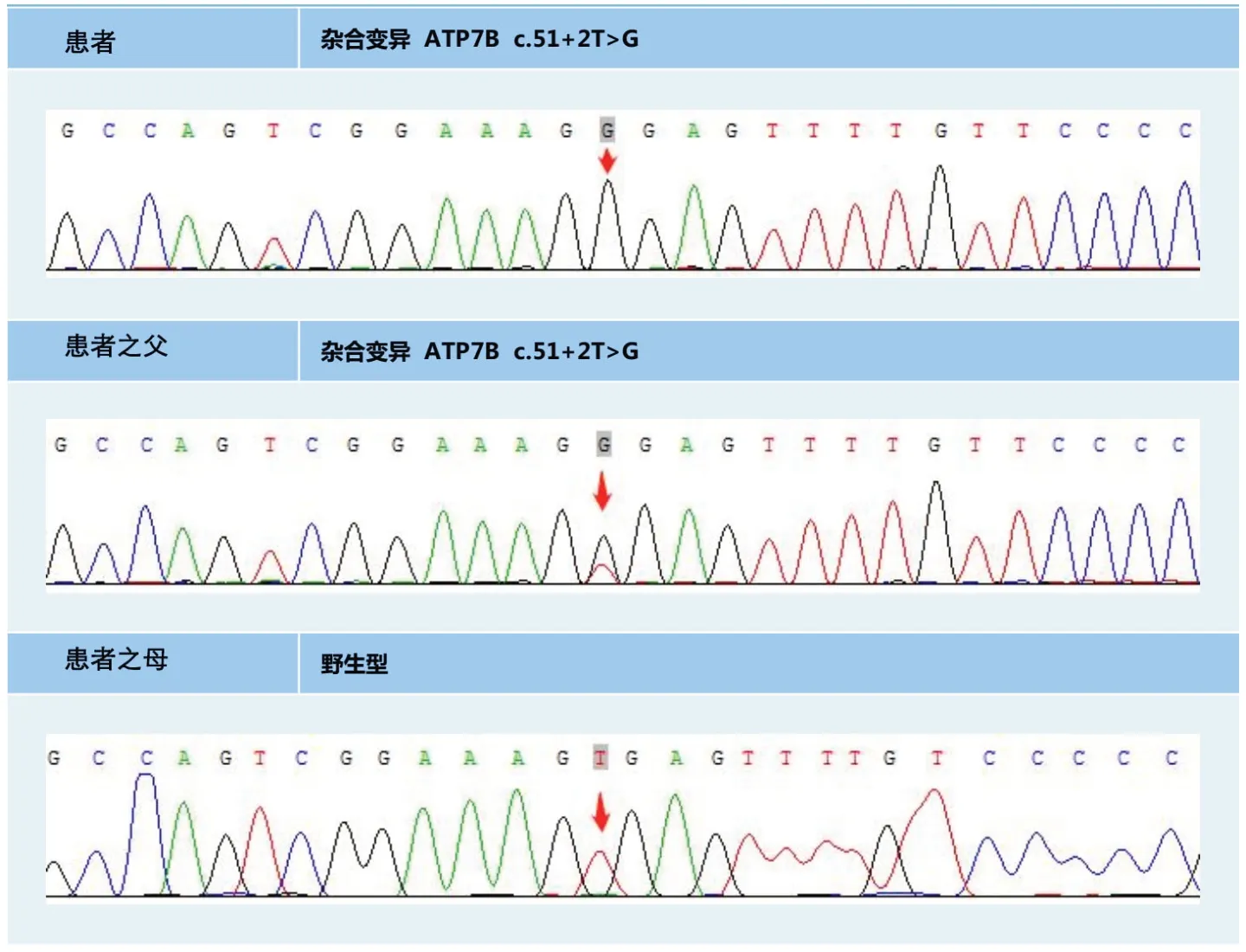
图1 患者及父母基因测序
2 讨论
卟啉病是由于先天性酶缺乏引起的卟啉代谢障碍,致卟啉及卟啉前体的产生与排泄增加,并积聚在组织中而发病。为常染色体显性遗传,主要临床特点包括暴露部位皮肤损害、急性腹痛、神经精神症状。按照卟啉生成的部位可以分为红细胞生成性卟啉病及肝卟啉病,根据临床表现可分为仅有皮肤光敏症状的卟啉病、仅有神经症状的急性卟啉病和神经症状和皮肤光敏症状共存的急性卟啉病三大类。肝卟啉病临床表现多不典型,呈多样化,特异性确诊检查少且多易呈阴性结果,临床误诊率高[2-3]。
本例患者以肝损害为突出表现,入院后进一步排除常见嗜肝病毒感染,自免肝全套未见异常,无导致心源性肝硬化的慢性循环系统疾病,腹部超声排除了布加综合征等导致肝硬化的常见血管性原因。但是该患者年仅18 周岁,却有明显肝脏肿大、典型肝硬化表现,提示病程可能偏长,需要考虑遗传代谢性疾病。结合该患者年轻女性,产后1 个月起病,以暴露部位皮肤损害、急性腹痛、黄疸为主要临床表现,需要考虑卟啉病的可能。根据《罕见病诊疗指南(2019 年版)》和《实用内科学》,其中指出无论混合型卟啉病患者还是遗传性粪卟啉病患者,其粪便中原卟啉和粪卟啉明显增高,发作时尿氨基酮戊酸和卟胆原排出增加,血清转氨酶升高少见,仅见于13%的急性发作期患者和一些临床无症状的患者。对照本例患者,检查结果不满足诊断标准。更重要的作为罕见遗传病,基因测序可明确具体突变,从而确定卟啉病类型。而该患者基因测序,未发现卟啉病相关基因突变,因而可以排除卟啉病诊断。
肝豆状核变性又称威尔逊病,是1912 年由Wilson 首先提出。本病是一种常染色体隐性遗传的铜代谢障碍疾病,致病基因是ATP7B,位于13q14.3,编码铜转运蛋白-P 型ATP 酶,该酶参与铜蓝蛋白的合成和铜的代谢。肝豆状核变性在世界范围内发病率1/30 000~1/100 000,致病基因携带者大约为1/90[4],ATP7B的缺失或功能异常导致铜在多个组织或者脏器积累,造成靶器官损害。临床上主要分为肝型、脑型以及其他类型,其主要临床表现为肝脏损伤、神经系统异常如构音障碍、肌张力障碍、震颤、帕金森征、舞蹈病、共济失调等及Coomb's 阴性溶血性贫血和眼部的K-F环等。WD 还可以表现为精神病症状,表现为性格改变,易冲动,情绪不稳定,性癖,和不得体的行为。既往一项回顾性研究表示51.04%(516/1011)被误诊为100 多种不同的疾病,另外还有19.09%(193/1011)未被诊断为特定疾病[5]。发病年龄多在5~35 岁。WD 常见检查包括血清铜蓝蛋白、尿铜、肝铜以及基因检测。在WD 中,铜蓝蛋白可降低;24 h 尿铜成人患者大于100 μg,儿童患者大于40 μg。基因检测可确定染色体突变,明确诊断[1,6-7]。WD 是少数能有效控制的遗传病之一,目前的治疗策略主要有:①利用D-青霉胺,二巯基丙磺酸钠和曲安汀等螯合剂增加尿中铜的排泄量;②利用锌盐抑制铜在消化道中的吸收;③肝移植可恢复肝脏功能并减轻门静脉高压症,仅适用于急性肝功能衰竭或终末期肝病无法通过药物治疗的患者[8-9]。WD 若能早期诊断,大部分患者预后良好。我国《肝豆状核变性诊断与治疗指南》[10]中的诊断要点包括:患者具有锥体外系症状或肝病表现;K-F 环阳性;血清铜蓝蛋白低于0.2 g/L;24 h 尿铜>100 μg,可临床诊断为肝豆状核变性。进一步ATP7B基因突变检测,发现两个等位基因致病突变具有确诊价值。也可参照2001 德国莱比锡第8 次WD 国际会议上制定的WD 评分系统(Leipzig),该评分全面系统地纳入了WD 的临床表现、生化检验和遗传分子学检测结果。该患者有肝病表现;K-F 环阳性;血清铜蓝蛋白66 mg/L;24 h 尿铜2 921.5 μg 符合临床诊断标准。ATP7B基因检测存在“ATP7B(13q14.3|NM_000053.3)Intron1 c.51+2T>G 剪接突变;高通量结果提示伴有chr13q14.3(52158814-53624916)区域内可能存在片段的新发杂合缺失,长度约1.47 Mb,包含ATP7B基因”。其患者父亲、妹妹基因检测均发现“ATP7B(13q14.3|NM_000053.3)Intron1 c.51+2T>G 剪接突变”,儿子基因测序发现“chr13q14.3(52158814-53624916)区域内可能存在片段的新发杂合缺失,长度约1.47 Mb,包含ATP7B基因”,也为诊断肝豆状核变性提供了线索,见图2。

图2 患者家系图
全世界范围内已报道ATP7B突变超过500 种,最常见的突变是点突变,其他类型的突变则很少见。在中国最常见的基因突变为p.R778L[11]。本例报道患者致病性突变c.51+2T>G 和杂合缺失是先证者两个染色体上的复合杂合子,分别从她的父亲继承和自身新发突变。结合文献研究发现有部分纯合子的患者中发现了完整的基因缺失,纯合子突变的患者要考虑是潜在的半合子状态[12],因此本例患者有发病可能。c.51+2T>G 突变被鉴定为ATP7B基因序列中的一个新的剪接突变,首次报道于2019 年。该突变是位于内含子1 的突变,导致外显子跳读而使所编码的蛋白质发生紊乱而丧失其正常功能[12]。由此导致患者无法编码ATP7B相应酶,进而铜积累发病。c.51+2T>G 该基因突变发现较晚,可能需要更多的研究来发现其该基因的作用机制。结合文献和笔者的这例患者表现表明受影响外显子两侧的内含子缺失可引起外显子跳读、内含子保留或其他基因的序列插入,内含子缺失可能是一种致病突变[12]。此外本例患者有特殊的皮肤光敏性损伤表现,可能是该基因突变的特殊表型。查阅文献仅发现1 例类似患者,表现为面、颈、双手暴露部位皮肤痛觉过敏,温热觉正常,无皮疹、硬肿及色素沉着,但该患者无肝病表现,且未行基因测序[13]。
综上所述,通过本例病例的报道和国内外文献的综述,可以帮助临床医生增强对本病的认识,提高对其临床症状的鉴别和诊断。本病的治疗效果与早期诊治关系密切,临床医生在临床诊治过程中需仔细询问病史,拓展思维,完善检查,减少或避免该病的误诊及漏诊。
