头花蓼质量标准的研究
2020-05-12赵英魁黄豆豆王璇江圣圭孙连娜
赵英魁黄豆豆王 璇江圣圭孙连娜∗
(1.海军军医大学药学院,上海 200433;2.上海中医药大学中药学院,上海 201203;3.福建中医药大学药学院,福建 福州 350108)
头花蓼又名太阳草、石莽草、水绣球等,为蓼科蓼属植物头花蓼Polygonum capitatumBuch.-Ham.ex D.Don 的干燥全草或地上部分,是少数民族常用药。在《广西中药志》 《贵州省中药材、民族药材质量标准》[1]《中国药用植物志》 《云南中草药》 《中药大辞典》 《中华本草》[2]等书籍中均有记载。全草入药,味苦、辛,性凉。头花蓼含有黄酮类[3-4]、酚酸类[5-7]、木脂素类[8]等,具有抗菌[9-10]、抗炎镇痛、抗氧化[11]、降糖[8]等药理作用。主要用于清热解毒、利尿通淋、肾盂肾炎、尿路结石、膀胱炎、风湿痛、跌打损伤、尿道感染、疮疡湿疹等症。目前,头花蓼被收载于2003 年版《贵州省中药材、民族药材质量标准》、2009 年版《湖南省中药材标准》。在两者地标中只规定了性状、TLC 鉴别、水分检查、含有量测定等项目,相关标准不够全面,且含有量测定项中只以槲皮素为质量控制指标,供试品溶液制备了盐酸水解方法。以头花蓼为原料的单方制剂热淋清颗粒[12]收载于2015 年版《中国药典》 的含有量测定项以没食子酸为对照品。课题组参考相关文献[13-19],选择代表性活性成分没食子酸和槲皮苷,运用显微鉴别法、TLC 法和HPLC 法对23 批药材进行了定性鉴别和含有量测定,并对药材的水分、总灰分、酸不溶性灰分等进行检查,初步制定检测限度,对其质量标准进行具体研究,以期为完善该药材的质量控制提供依据。
1 材料
Waters 1525 高效液相色谱仪(美国沃特世公司);Sartorius CPA225D 电子分析天平(赛多利斯科学仪器北京有限公司);KQ-500 型超声仪(昆山市超声仪器有限公司);Biomate 3S UV-visible spectrophotometer(美国赛默飞世尔科技公司)。
正相硅胶HPTLC Silica gel 60(德国默克公司,货号1.05641.0001,批号HX257461)。乙腈(色谱纯)、水为蒸馏水、其他试剂均为分析纯。没食子酸对照品(批号110831-201204)、槲皮苷对照品(批号111538-200504)均购于中国食品药品检定研究院。
头花蓼药材对照品及23 批头花蓼药材,由海军军医大学陈万生教授鉴定为正品,信息见表1。
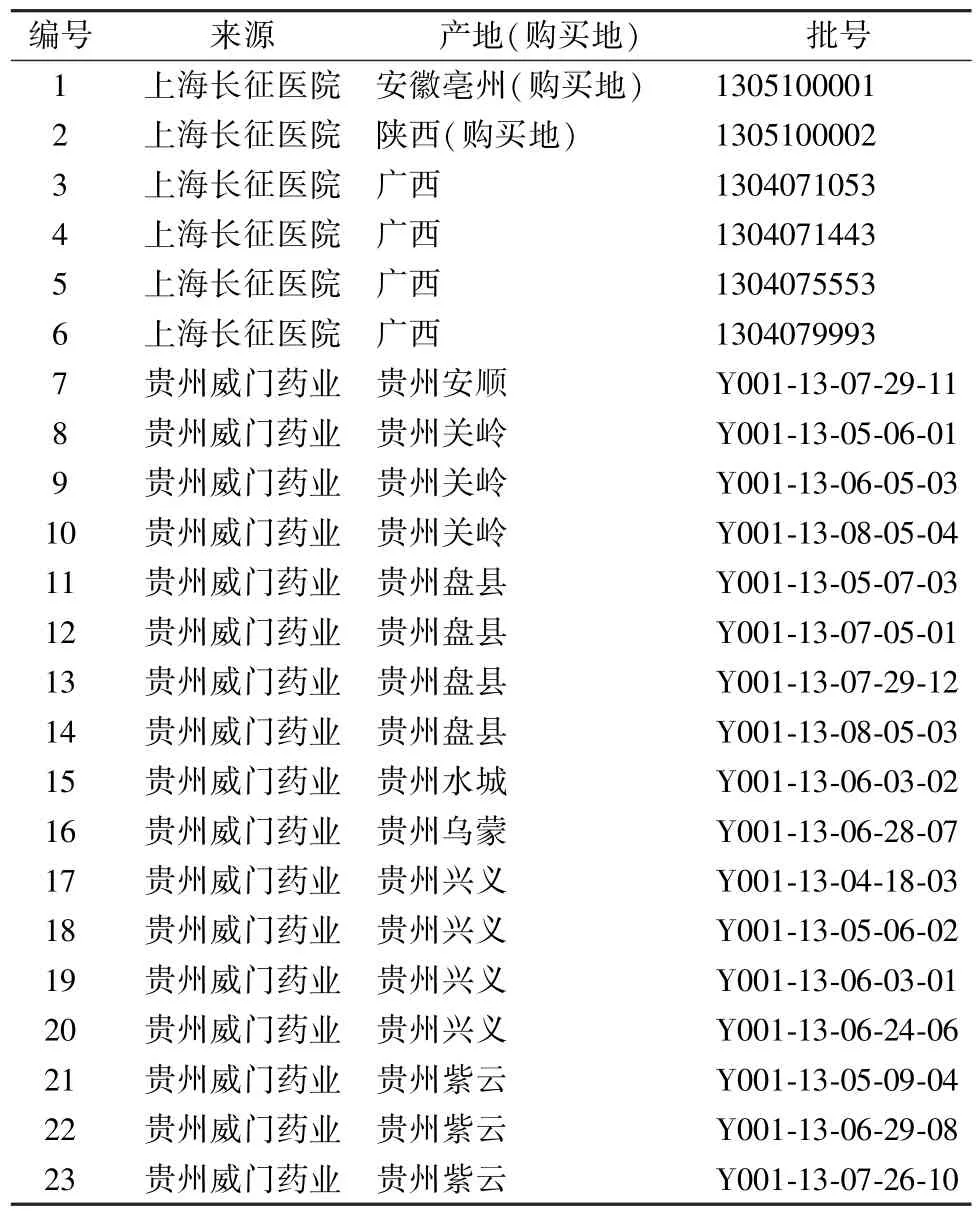
表1 样品信息Tab.1 Information of samples
2 方法与结果
2.1 显微鉴别 对头花蓼药材叶的表面观(临时水装片)及药材粉末(水合氯醛装片)的相关显微特征进行鉴别观察。叶的表面观,上表皮细胞垂周壁近平直,下表皮细胞垂周壁略呈波状弯曲。气孔不定式,副卫细胞3~4 个,上下表皮均有分布,下表皮较多。腺毛可见,常含有红棕色物质,腺柄由1~3 列细胞组成,细胞壁明显增厚,并具疣状突起。粉末特征,本品粉末棕褐色,草酸钙簇晶多,大小悬殊,直径10~75 μm。纤维多成束或散在,易断裂,多壁厚,具明显孔沟。可见螺纹及环纹导管。淀粉粒众多,常见单粒,类圆形、卵圆形等。显微特征见图1。

图1 头花蓼叶表面和药材粉末显微特征图Fig.1 Micropic characteristics graph of leaf surface of P. capitatum and medicinal materials powder
2.2 TLC 定性鉴别
2.2.1 展开体系优化 考察二氯甲烷-乙酸乙酯-甲酸(2∶8∶1)、正己烷-乙酸乙酯-甲酸(2∶8∶0.5)、甲苯-乙酸乙酯-甲酸(2∶6∶1.5)、甲苯-乙酸乙酯-甲酸(2∶8∶1)、甲苯-乙酸乙酯-甲酸(2∶10∶1)、甲醇-水-甲酸(9∶5∶5)等展开体系,结果发现在甲苯-乙酸乙酯-甲酸(2∶8∶1)体系下,斑点清晰,分离度好。
2.2.2 载体考察 在甲苯-乙酸乙酯-甲酸(2∶8∶1)体系下,考察硅胶G 预制板、聚酰胺薄膜、高效硅胶G 预制板3 种载体,结果发现高效硅胶G预制板为载体时,斑点分离完全,较清晰,展开结果理想。
2.2.3 耐用性考察 考察不同品牌薄层板[银龙牌的硅胶HSG 预制板(批号20130531)、黄海牌硅胶HSG 预制板(批号20130601)、默克正相硅胶HPTLC Silica gel 60(货号1.05641.0001,批号HX257461)];不同温度(4、12、22 ℃);不同湿度(32%、52%、72%),发现硅胶G 板、温度、湿度对结果影响不大,展开结果均较好,表明该方法耐用性良好。
2.2.3 方法与结果 取头花蓼粉末1 g,置50 mL具塞锥形瓶中,加20% 甲醇20 mL,超声处理30 min,取出,滤过,滤液蒸干,残渣用甲醇定容至1 mL 量瓶中,作为供试品溶液。精密称取适量没食子酸、槲皮苷对照品,加甲醇分别制成每1 mL含1.0、0.5 mg 溶液,作为对照品溶液。另取头花蓼对照药材1 g,同法制备对照药材溶液。照TLC 2015 年版《中国药典》 四部0502 试验,吸取上述供试品和对照药材溶液3 μL、对照品溶液各2 μL,分别点于同一高效硅胶G 薄层板上,以甲苯-乙酸乙酯-甲酸(2∶8∶1)为展开剂,展开,取出,晾干,喷以5%三氯化铝乙醇溶液,热风吹至斑点显色清晰,置紫外光灯(365 nm)下检视,结果见图2。供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同颜色的荧光斑点,斑点清晰,分离度好。

图2 头花蓼TLC 图Fig.2 TLC chromatograms of P. capitatum
2.3 水分、总灰分、酸不溶性灰分、浸出物检查 根据2015 年版《中国药典》 四部方法测定,结果见表2。
2.4 没食子酸含有量测定
2.4.1 波长选择 将没食子酸对照品溶液,在200~700 nm 范围内扫描,发现没食子酸在220、273 nm 波长处有最大吸收,按《中国药典》 有关规定,先检查了所使用乙腈溶剂在供试品测定所用波长附近符合要求,结合其他相关文献,最后选择270 nm 作为没食子酸的检测波长。
2.4.2 流动相选择 参照2015 年版《中国药典》一部及相关文献,广枣、余甘子、蓝布正等文献资料中对没食子酸含有量测定的条件,考察了甲醇-0.2%磷酸(18∶82)、乙腈-0.2% 甲酸(5∶95)、乙腈-0.2%磷酸(5∶95)对没食子酸的洗脱效果,结果发现乙腈-0.2%磷酸(5∶95)流动相下,没食子酸的峰型较好,与周边峰无相互干扰,达到完全分离。故选择选择乙腈-0.2%磷酸(5∶95)作为流动相。
2.4.3 色谱条件 迪马Dikma Diamonsil C18色谱柱(4.6 mm×250 mm,5 μm);流动相乙腈-0.2% 磷酸溶液(5∶95);检测波长270 nm;体积流量1 mL/min;进样量20 μL。
2.4.4 对照品溶液制备 精密称取没食子酸对照品5.52 mg,置10 mL 量瓶中,加甲醇溶解定容摇匀,得质量浓度为0.552 mg/mL 的对照品贮备液。
2.4.5 供试品溶液制备 取本品粉末(过3 号筛)约0.5 g,精密称定,置50 mL 具塞锥形瓶中,精密加入10%甲醇25 mL,称定质量,置水浴中加热回流60 min,放冷,称定质量,用10%甲醇补足减失质量,摇匀,滤过,取续滤液,即得。

表2 水分、总灰分、酸不溶性灰分、浸出物含有量测定结果Tab.2 Results of content determination of water,total ash,acid-insoluble ash and ectract
2.4.6 系统适用性试验 在“2.4.1”项色谱条件下,分别取没食子酸对照品溶液、供试品溶液注入液相色谱仪,没食子酸的保留时间8.2 min,没食子酸和其相近的其他峰分离完全(分离度>1.5),即本实验条件下没食子酸与其他组分分离完全。理论板数以没食子酸计算为3 000,色谱图见图3。
2.4.7 线性关系考察 将没食子酸对照品贮备液用甲醇 稀释制成 552、276、138、69、34.5、17.25、8.626 μg/mL 的系列溶液,在“2.4.1”项色谱条件下测定,记录峰面积,以峰面积为纵坐标(Y),没食子酸质量为横坐标(X)进行回归,得回归方程为Y=2 959 114.4X+67 276.7,r=0.999 9,表明没食子酸在0.1725~11.04 g 范围内线性关系良好。
2.4.8 精密度试验 精密吸取没食子酸对照品溶液20 μL,在“2.4.1”项色谱条件下,重复进样6次,测得峰面积RSD 为0.92%,表明仪器精密度良好。
2.4.9 稳定性试验 精密吸取1 号药材供试品溶液20 μL,在“2.4.1”项色谱条件下,分别于0、4、8、12、24 h 进样,测得峰面积RSD 为1.90%,表明供试品溶液在24 h 内稳定性良好。

图3 没食子酸HPLC 色谱图Fig.3 HPLC chromatograms of gallic acid
2.4.10 重复性试验 取1 号药材,平行制备6 份供试品溶液,在“2.4.1”项色谱条件下,分别进样20 μL,记录峰面积。得没食子酸平均含有量为1.614 1 mg/g,RSD 为1.67%,表明该方法重复性良好。
2.4.11 加样回收率试验 精密称定1 号样品(没食子酸含有量1.614 1 mg/g)约0.25 g,共6 份,置于具塞锥形瓶中,精密加入样品含有量100%的没食子酸对照品,按“2.4.3”项下方法制备供试品溶液,在“2.4.1”项色谱条件下测定,得没食子酸平均加样回收率为98.90%,RSD 为1.90%,结果见表4。
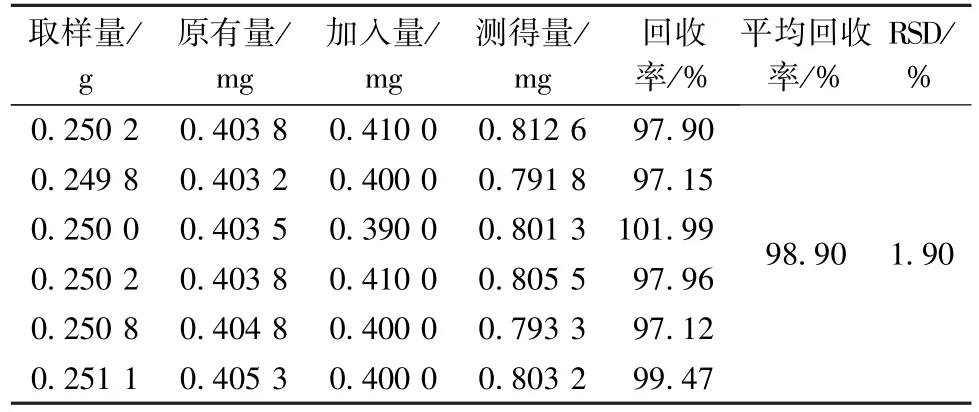
表4 没食子酸加样回收率试验结果(n=6)Tab.4 Results of recovery tests for gallic acid(n=6)
2.4.12 样品含有量测定 分别取上述23 批药材,按“2.4.3”项下方 法制备 供试品溶液,在“2.4.1”项色谱条件下测定,结果见表5。
2.5 槲皮苷含有量测定
2.5.1 波长选择 将槲皮苷对照品溶液,在200~700 nm 范围内扫描,发现槲皮苷在257、348 nm波长处有最大吸收,按《中国药典》 有关规定,先检查了所使用的乙腈溶剂在供试品测定所用波长附近是符合要求的,结合其他相关文献,最后选择254 nm 分别作为槲皮苷的检测波长。

表5 没食子酸含有量测定结果(n=3,mg/g)Tab.5 Results of content determination of gallic acid(n=3,mg/g)
2.5.2 流动相选择 参照2015 年版《中国药典》一部及相关文献,侧柏叶、合欢花等文献资料中对槲皮苷含有量测定的条件,考察了甲醇-0.2%磷酸(48∶ 52)、甲醇-0.2% 磷酸(46∶ 54)、乙 腈-0.2%甲酸(21∶79)、乙腈-0.2%磷酸(25∶75)、乙腈-0.2%磷酸(5∶95)对槲皮苷的洗脱效果,结果发现乙腈-0.2%磷酸(5∶95)流动相下,槲皮苷的峰型较好,与周边峰无相互干扰,达到完全分离。故选择乙腈-0.2% 磷酸(5∶95)作为流动相。
2.5.3 色谱条件 迪马Dikma DiamonsiL C18色谱柱(4.6 mm×250 mm,5 μm);流动相乙腈-0.2%磷酸溶液(21∶79);检测波长254 nm;体积流量1 mL/min;进样量20 μL。
2.5.4 对照品溶液制备 精密称取槲皮苷对照品6.26 mg,置10 mL 量瓶中,加甲醇溶解定容摇匀,即得质量浓度为0.626 mg/mL 的对照品贮备液。
2.5.5 供试品溶液制备 取本品粉末(过3 号筛)约0.5 g,精密称定,置50 mL 具塞锥形瓶中,精密加入80%甲醇25 mL,称定质量,置水浴中加热回流60 min,放冷,称定质量,用80%甲醇补足减失质量,摇匀,滤过,取续滤液,即得。
2.5.6 系统适用性试验 在“2.5.1”色谱条件下,分别取槲皮苷对照品溶液、供试品溶液注入液相色谱仪,槲皮苷的保留时间14.3 min,槲皮苷和其相近的其他峰分离完全(分离度>1.5),即本实验条件下槲皮苷与其他组分分离完全。理论板数以槲皮苷计算为3 000,色谱图见图4。

图4 槲皮苷HPLC 色谱图Fig.4 HPLC chromatograms of quercitin
2.5.7 线性关系考察 将没槲皮苷对照品贮备液用甲醇 稀释制成 626、313、156.5、78.25、39.125、19.562 5、9.781 25 μg/mL 的系列溶液,分别吸取20 μL 注入液相色谱仪,记录色谱图,以峰面积为纵坐标(Y),没食子酸质量为横坐标(X)进行回归,得回归方程为Y=2 537 136.6X+34 562.2,r=0.999 9,表明没槲皮苷在0.195 6~12.52 g 范围内线性关系良好。
2.5.8 精密度试验 精密吸取槲皮苷对照品溶液20 μL,在“2.5.1”色谱条件下,重复进样6 次,测得峰面积RSD 为0.86%,表明仪器精密度良好。
2.5.9 稳定性试验 精密吸取1 号药材供试品溶液20 μL,在“2.5.1”色谱条件下,分别于0、4、8、12、24 h 进样,测得峰面积RSD 为1.24%,表明供试品溶液在24 h 内稳定性良好。
2.5.10 重复性试验 取1 号药材,平行制备6 份供试品溶液,在“2.5.1”色谱条件下,分别进样20 μL,记录峰面积。得槲皮苷均含有量为3.891 8 mg/g,RSD为1.50%,表明该方法重复性良好。
2.5.11 加样回收率试验 精密称定1 号样品(槲皮苷含有量3.891 8 mg/g)约0.25 g,共6 份,置于具塞锥形瓶中,精密加入样品含有量100%的槲皮苷对照品,按“2.5.3”项下方法制备供试品溶液,在“2.5.1”色谱条件下进样,得槲皮苷平均加样回收率为98.89%,RSD 为3.05%,结果见表6。
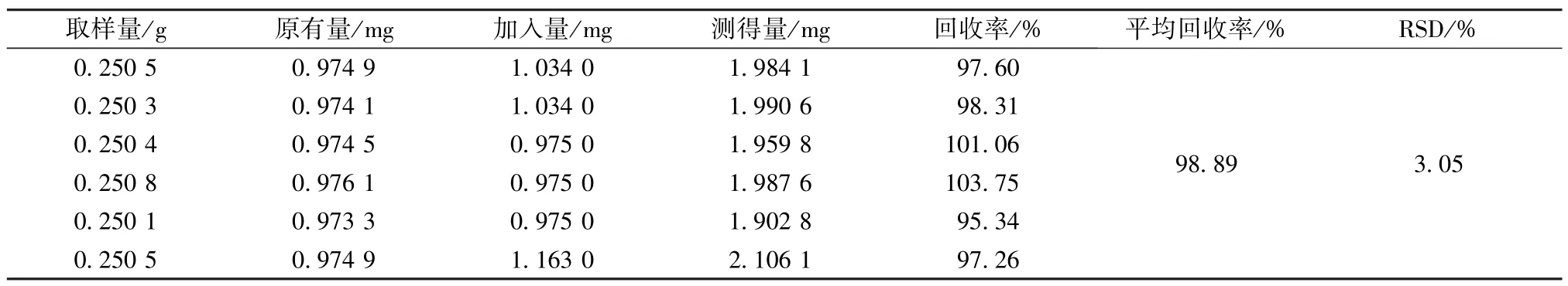
表6 槲皮苷加样回收率试验结果(n=6)Tab.6 Results of recovery tests for quercitin(n=6)
2.5.12 样品含有量测定 分别取上述23 批药材,按“2.5.3”项下方 法制备 供试品溶液,在“2.5.1”色谱条件下进样,结果见表7。

表7 槲皮苷含有量测定结果(n=3,mg/g)Tab.7 Results of content determination of quercitin(n =3,mg/g)
3 结果与讨论
3.1 检查与浸出物限量 本实验发现,23 批药材水分范围为7.75%~11.63%,平均为9.75%,暂定水分不得超过12.0%;总灰分范围4.04%~18.79%,均值为11.47%,暂定总灰分不得超过14.0%;酸不溶性灰分范围0.14%~8.62%,均值为2.62%,暂定酸不溶性灰分不得超过4.0%;50%乙醇浸出物范围15.49%~25.26%,均值为20.50%,暂定不得低于15.0%。
3.2 没食子酸、槲皮苷含有量和限量 实验结果表明23 批样品中没食子酸、槲皮苷含有量差异较大,即使同一地区含有量也不尽相同。没食子酸以陕西购的(2 号)含有量最高,贵州盘县产地(14号)含有量最低;槲皮苷以贵州盘县产地(13号)含有量最高,广西产地(6 号)含有量最低。根据结果,暂定以干燥品计,没食子酸含有量不得少于0.05%、槲皮苷含有量不得少于0.10%。
通过本研究,课题组完善了头花蓼质量标准的研究,与贵州、湖南两者地标相比,增加了显微鉴别项,总灰分、酸不溶性灰分、50%乙醇浸出物检查项,选择代表性活性成分没食子酸和槲皮苷作为质控指标,以期为制定头花蓼质量标准提供依据,为完善药材的质量控制控制奠定基础。
