3 种淫羊藿苷次生产物的制备及HPLC 测定
2020-03-27杨轶舜张越陈嘉雯周奕嘉赵日吉吴华燕宋泽家
杨轶舜张 彤∗丁 越陈嘉雯周奕嘉赵日吉吴华燕宋泽家
(1.上海中医药大学教学实验中心,上海201203; 2.上海中医药大学中药学院,上海201203)
淫羊藿,又名仙灵脾,其主要功效为补肝肾、强筋骨[1]。淫羊藿苷为淫羊藿主要有效成分。近年来,关于中药有效成分体内代谢研究表明,中药有效成分除了以原型化合物发挥作用外,还有一部分需经过肠内菌群的作用,转化为代谢产物后才能发挥药效作用[2]。宝藿苷Ⅰ(淫羊藿次苷Ⅱ)和脱水淫羊藿素(淫羊藿苷元)是淫羊藿苷的次生苷和苷元,也是其主要代谢产物,即淫羊藿苷脱去一分子葡萄糖后生成宝藿苷Ⅰ,再脱去一分子鼠李糖生成脱水淫羊藿素(淫羊藿苷元)。王婷等[3]对6 种淫羊藿中的黄酮类成分进行了抗肿瘤活性的研究,结果表明淫羊藿苷、木犀草素、朝藿素和宝藿苷Ⅰ对人乳腺癌细胞MCF-7 和人肝癌细胞HepG2 的增殖有抑制作用,且宝藿苷Ⅰ对MCF-7细胞的抑制效果强于淫羊藿苷。宝藿苷Ⅰ有较好的抗肺癌药效作用,其药效优于淫羊藿中其他黄酮类多糖苷[4]。药理研究表明,脱水淫羊藿素有广谱的抗肿瘤作用,其对人肝脏、血液淋巴系统、乳腺、结肠、胰腺、肺、前列腺、宫颈等肿瘤细胞具有明显的抑制作用[5]。此外,淫羊藿次苷Ⅰ是淫羊藿苷脱去一分子鼠李糖生成的次生苷,其抗骨质疏松活性优于淫羊藿苷[6]。
目前,淫羊藿苷次生苷和苷元的制备方法主要有酸水解法、酶水解法和酸解-酶解两步法[5]。酸水解法采用盐酸、硫酸或硫酸与醋酸混合的方法水解淫羊藿苷[7],但酸水解反应条件剧烈,易生成副产物。而酶水解反应具有特异性强、反应条件温和、产率高、分离纯化简单、对环境没有污染等优点[8]。常用于水解淫羊藿苷的酶为β-葡萄糖苷酶、纤维素酶、柚皮苷酶和蜗牛酶。由于淫羊藿苷含有2 个糖基,因此不同酶水解生成的产物不同。研究表明,β-葡萄糖苷酶[9]、纤维素酶[10]及柚皮苷酶均能水解淫羊藿苷生成宝藿苷Ⅰ,且β-葡萄糖苷酶的转化率显著高于其他2 种酶[4]。另一方面,柚皮苷酶和蜗牛酶能水解淫羊藿苷生成宝藿苷Ⅰ和脱水淫羊藿素[5]。但鲜有淫羊藿次苷Ⅰ制备方法的报道,尤其是酶解法往往无法制备淫羊藿次苷Ⅰ,能够水解α-鼠李糖苷键的酶如柚皮苷酶和蜗牛酶会同时水解β-葡萄糖苷键,在此类酶的作用下往往生成宝藿苷Ⅰ和脱水淫羊藿素,无法生成淫羊藿次苷Ⅰ。易鹏等[11]研究表明,淫羊藿苷的葡萄糖苷键容易被β-葡萄糖苷酶水解,而其鼠李糖苷键易被稀硫酸水解,所以可以采用酸解和酶解2 种方法相结合来制备脱水淫羊藿素。
本研究采用酸解法制备淫羊藿次苷Ⅰ、酶解法制备宝藿苷Ⅰ,以及酸解和酶解结合法制备脱水淫羊藿素,并建立HPLC 法同时测定3 种淫羊藿苷次生产物的含有量。
1 仪器与试药
Aglient 1260 型高效液相色谱仪(美国安捷伦公司);PH 计(美国Eutech 公司);XS105 型微量分析天平(瑞士梅特勒-托利多公司);DF-101S 型恒温磁力搅拌器(上海鹰迪仪器设备有限公司)。淫羊藿次苷Ⅰ(批号YJ06285A14)、宝藿苷Ⅰ(批号HS0918XA13)、脱水淫羊藿素(淫羊藿苷元,批号YA0903S13)对照品和蜗牛酶均购自上海源叶生物科技有限公司;淫羊藿苷(武汉远成共创科技有限公司);β-葡萄糖苷酶(实验室自制,来源于里氏木霉QM9414)。乙腈和甲醇为色谱纯(安徽时联特种溶剂公司);其他试剂均为分析纯或生化纯(国药集团化学试剂有限公司);水为超纯水。
2 方法与结果
2.1 淫羊藿次苷Ⅰ制备 称取淫羊藿苷50 mg,加入乙醇和5%稀硫酸溶液各3.75 mL,50 ℃搅拌反应24 h。抽滤反应液,收集沉淀。沉淀在40 ℃下真空干燥3 h,得淫羊藿次苷Ⅰ粗品,硅胶柱层析,得精制品。
2.2 宝藿苷Ⅰ制备 称取淫羊藿苷和β-葡萄糖苷酶各50 mg,加入0.2 mol/L pH 4.0 的醋酸-醋酸钠缓冲液10 mL,40 ℃搅拌反应5 h。乙酸乙酯萃取(50 mL×3 次),收集有机相,用少量无水硫酸钠干燥,减压浓缩,浓缩液转移至4 ℃冰箱静置结晶。收集结晶,在40 ℃下真空干燥3 h,得宝藿苷Ⅰ粗品,将粗品用甲醇重结晶,得精制品。
2.3 淫羊藿苷元(脱水淫羊藿素)制备
2.3.1 蜗牛酶解法 称取淫羊藿苷和蜗牛酶各20 mg 于20 mL磷酸缓冲液(0.2 mol/L,pH 6.0)中,37 ℃搅拌5 h,乙酸乙酯萃取(20 mL×3 次),收集上层溶液,用少量无水硫酸钠干燥,旋蒸浓缩,浓缩液转移至4 ℃冰箱静置结晶。结晶在40 ℃下真空干燥3 h,成品于4 ℃密封保存。
2.3.2 酸解-酶解法 称取淫羊藿苷50 mg,依次加入乙醇和5%稀硫酸溶液各3.75 mL,在50 ℃水浴中搅拌24 h,抽滤,收集沉淀Ⅰ(淫羊藿次苷Ⅰ)。将50 mg β-葡萄糖苷酶溶于10 mL 0.2 mol/L pH 4.0 的醋酸缓冲液中,将得到的沉淀Ⅰ充分转移至酶溶液中,置于40 ℃水浴中继续搅拌8 h,抽滤反应液,收集沉淀Ⅱ(脱水淫羊藿素),滤液用乙醚萃取(15 mL×3 次),收集有机相,用少量无水硫酸钠干燥,减压浓缩,浓缩液转移至4 ℃冰箱静置结晶。将结晶和沉淀Ⅱ在40 ℃下真空干燥3 h,得脱水淫羊藿素粗品,硅胶柱层析,得精制品。
2.3.3 酶解-酸解法 称取β-葡萄糖苷酶50 mg 于10 mL 0.2 mol/L pH 4.0 的醋酸缓冲液中,待酶溶解后,取50 mg淫羊藿苷置于酶溶液中,于40 ℃水浴中搅拌8 h,加入乙醇和5%稀硫酸溶液各3.75 mL,在50 ℃水浴中继续搅拌24 h,抽滤,保留沉淀,再将滤液用乙醚萃取(15 mL×3次),保留上层溶液,用少量无水硫酸钠干燥,旋蒸浓缩,浓缩液转移至4 ℃冰箱静置结晶。将结晶和沉淀在40 ℃下真空干燥3 h,成品于4 ℃密封保存。
2.3.4 混酸法 称取28 mg 淫羊藿苷,加入0.6 mL 5 mol/L硫酸溶液和2 mL 80%冰醋酸,于80 ℃水浴中回流搅拌24 h,乙醚萃取(10 mL×3 次),保留上层溶液,用少量无水硫酸钠干燥,旋蒸浓缩,浓缩液转移至4 ℃冰箱静置结晶。结晶在40 ℃下真空干燥3 h,成品于4 ℃密封保存。
2.4 淫羊藿苷次生产物测定
2.4.1 色谱条件 Diamonsil C18色谱柱(250 mm×4.6 mm,5 μm);流动相乙腈-0.1% 磷酸(70∶ 30);体积流量1 mL/min;进样量10 μL;检测波长270 nm;柱温30 ℃。色谱图见图1。
2.4.2 溶液制备
2.4.2.1 对照品溶液 分别精密称取淫羊藿次苷Ⅰ、宝藿苷Ⅰ、脱水淫羊藿素对照品7.44、9.66、10.24 mg 于10 mL量瓶中,乙腈-0.1%磷酸(70∶30)溶解后定容,摇匀,配制成淫羊藿次苷Ⅰ、宝藿苷Ⅰ、脱水淫羊藿素质量浓度分别为744、966、1 024 μg/mL 的溶液,即得。
2.4.2.2 供试品溶液 精密称取按“2.1”“2.2”“2.3”项下方法制备的淫羊藿苷各水解产物5 mg,分别置于10 mL量瓶中,加入乙腈-0.1%磷酸(70∶30)溶解后,稀释定容,过0.45 μm 微孔滤膜,即得。
2.4.3 线性关系考察 将“2.4.2.1”项下对照品溶液用乙腈-0.1%磷酸(70∶30)稀释2、4、10、20、40、100倍,即得系列质量浓度,在“2.4.1”项色谱条件下测定。以峰面积为纵坐标(Y),样品质量浓度为横坐标(X)进行回归,结果见表1,可知各成分在各自范围内线性关系良好。

表1 各成分线性关系
2.4.4 精密度试验 取“2.4.2.1”项下对照品溶液适量,在“2.4.1”项色谱条件下连续进样6 次,测得淫羊藿次苷Ⅰ、宝藿苷Ⅰ、脱水淫羊藿素峰面积RSD 分别为0.16%、0.21%、0.25%,表明仪器精密度良好。
2.4.5 重复性试验 按“2.4.2.2”项下方法制备供试品溶液6 份,在“2.4.1”项色谱条件下进样,测得淫羊藿次苷Ⅰ、宝藿苷Ⅰ、脱水淫羊藿素峰面积RSD 分别为1.64%、1.47%、1.51%,表明该方法重复性良好。
2.4.6 稳定性试验 取同一供试品溶液,于0、2、4、6、8、10、12、14、16、18 h 在“2.4.1”项色谱条件下进样,测得淫羊藿次苷Ⅰ、宝藿苷Ⅰ、脱水淫羊藿素峰面积RSD分别为1.92%、0.88%、0.49%,表明供试品在18 h内稳定性良好。

图1 各成分HPLC 色谱图
2.4.7 加样回收率试验 称取淫羊藿苷50 mg,依次加入乙醇和5% 稀硫酸溶液各3.75 mL,在50 ℃水浴中搅拌10 h,抽滤,收集沉淀Ⅰ。将50 mg β-葡萄糖苷酶溶于10 mL 0.2 mol/L pH 4.0 醋酸缓冲液中,将得到的沉淀Ⅰ充分转移至酶溶液中,置于40 ℃水浴中继续搅拌3 h,抽滤反应液,收集沉淀Ⅱ。沉淀Ⅱ在40 ℃下真空干燥3 h,得含有淫羊藿次苷Ⅰ、宝藿苷Ⅰ、脱水淫羊藿素的混合样品,并按“2.4.2.2”项下方法制备供试品溶液6 份,在“2.4.1”项色谱条件下进样,测混合样品中淫羊藿次苷Ⅰ、宝藿苷Ⅰ和脱水淫羊藿素的含有量。
取各成分已知的供试品溶液6 份,精密加入淫羊藿次苷Ⅰ、宝藿苷Ⅰ、脱水淫羊藿素对照品适量,在“2.4.1”项色谱条件下进样,测得淫羊藿次苷Ⅰ、宝藿苷Ⅰ、脱水淫羊藿素的平均加样回收率分别为98.63%、99.64%、100.07%,RSD 分别为0.53%、0.30%、0.64%。
2.4.8 样品含有量测定 按“2.1”项下方法酸水解得到淫羊藿次苷Ⅰ,其含有量为82.15%,产率为60.01%。按“2.2”项下方法通过酶水解淫羊藿苷得到的宝藿苷Ⅰ,其含有量为74.53%,产率为66.03%。
按“2.3”项下方法,制备脱水淫羊藿素。(1)酸解-酶解法,即先经过硫酸水解淫羊藿苷生成淫羊藿次苷Ⅰ,再用β-葡萄糖苷酶水解淫羊藿次苷Ⅰ生成脱水淫羊藿素;(2)酶解-酸解法,即先用β-葡萄糖苷酶水解淫羊藿苷生成宝藿苷Ⅰ,再用硫酸水解宝藿苷Ⅰ生成脱水淫羊藿素。结果表明,蜗牛酶水解法、酸解-酶解法和酶解-酸解法水解淫羊藿苷的反应产物中均有脱水淫羊藿素,但蜗牛酶水解法和酶解-酸解法产物中含有较多副产物、产率不高,分别为18.01%和21.84%,见表2;酸解-酶解法产物纯度和产率均较高,其含有量为88.01%,产率为45.61%;而硫酸-醋酸混合法(混酸法)所得产物并非脱水淫羊藿素,而是极性比其小的另一产物。

表2 各淫羊藿苷水解产物的含有量和产率
淫羊藿次苷Ⅰ粗品经硅胶柱层析后,含有量为99.78%,总得率为47.23%;宝藿苷Ⅰ粗品经重结晶后,含有量为98.43%,总得率为44.70%;将酸解-酶解法所得产物—脱水淫羊藿素粗品经硅胶柱层析后,含有量为99.38%,总得率为32.29%。
3 讨论
由于淫羊藿苷含有2 个糖基——葡萄糖基和鼠李糖基,见图2。因此,酸水解法常常会产生淫羊藿次苷Ⅰ、宝藿苷Ⅰ和脱水淫羊藿素等多种产物,难以获得单一的产物。目前,文献报道的酸水解法采用盐酸、硫酸或硫酸与醋酸混合的方法水解淫羊藿苷[5]。本研究表明,盐酸水解淫羊藿苷可以生成宝藿苷Ⅰ和脱水淫羊藿素,但有明显的副产物生成。硫酸水解淫羊藿苷可获得淫羊藿次苷Ⅰ,且其纯度高,粗品含有量为82.15%,产率为60.01%;β-葡萄糖苷酶水解法可获得宝藿苷Ⅰ,且纯度较高,粗品含有量为74.53%,产率为66.03%。
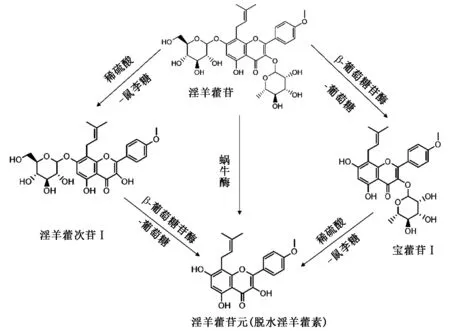
图2 淫羊藿苷次生产物制备路线图
虽有报道称硫酸与醋酸混合的方法水解淫羊藿苷可获得脱水淫羊藿素[7],但本研究表明,该方法无法获得脱水淫羊藿素,而是极性较小的另一化合物。蜗牛酶水解和酶解-酸解法虽可获得脱水淫羊藿素,但生成副产物较多导致产品纯度不高,产率低,蜗牛酶水解淫羊藿苷产率仅为18.01%,酶解-酸解法产率为21.84%;酸解-酶解法产物纯度和产率均较高,粗品含有量为88.01%,产率为45.61%,因此适用于脱水淫羊藿素的制备。
本实验建立了淫羊藿苷次生苷和苷元的制备方法,即通过酸解法制备淫羊藿次苷Ⅰ、酶解法制备宝藿苷Ⅰ,以及酸解和酶解结合法制备脱水淫羊藿素(淫羊藿苷元),并通过重结晶纯化获得了纯度大于98%的宝藿苷Ⅰ,通过硅胶柱层析纯化获得了纯度大于98%的淫羊藿次苷Ⅰ和脱水淫羊藿素。该方法操作简便、得率高,产物分离纯化步骤较简单,纯化产物纯度高,以期为开展淫羊藿苷次生产物药理活性研究奠定基础。
