基因编辑阳性细胞的富集报告系统
2020-02-06张力弦王启扉曹杨叶承宇陈智洋徐启杰邹秉杰宋沁馨周国华
张力弦 王启扉 曹杨 叶承宇 陈智洋 徐启杰 邹秉杰 宋沁馨 周国华
摘 要 基因编辑技术已成为医学生物学研究的重要工具,将不同基因快速高效地富集到基因编辑阳性细胞并进行针对性研究是促进基因编辑技术发展的关键。近年来,基于非同源末端接合、同源直接修复、单链退火、倒位重排等基因组修复机制,研究者利用荧光蛋白或抗性标签基因修复后表达的原理, 建立了多种可用于基因编辑阳性细胞筛选与富集的报告系统。富集后的细胞采用T7E1酶切法或测序技术进行突变分析, 呈现显著降低的背景信号和增加的突变比例。此类报告系统有利于基因编辑效果的表征。此外,分选后的阳性细胞可继续培养,在突变细胞系构建及突变细胞功能研究等领域具有良好的发展前景。本文对这些报告系统的设计原理与应用进行了总结,以期为建立更加完善的基因编辑评价系统提供参考。
关键词 基因编辑; 修复机制; 细胞富集; 报告系统; 评述
1 引 言
基因工程始于猴病毒SV40与大肠杆菌噬菌体λDNA首次拼接成环实验[1],随后,同源重组(Homologous recombination, HR)和非同源末端接合等基因修复机制以及核酸内切酶的发现与研究为现代基因编辑技术提供了理论基础[2,3]。近年来,研究人员发现了一些基于长序列识别的核酸酶,如大范围核酸酶(Meganuclease)[4~7]或归巢核酸内切酶(Homing endonuclease)、锌指核酸酶(Zinc finger nucleases,ZFNs)[8~12]、转录激活样效应物核酸酶(Transcription activator-like effector nucleases, TALENs)[13~17]、结构识别核酸酶(Structure-guided nuclease, SGN)[18],以及成簇规律间隔短回文重复相关核酸酶Cas9(Clustered regularly interspaced short palindromic repeats associated protein Cas9,CRISPR/Cas9)[19~22]。上述核酸酶可精确地识别靶序列并诱导双链断裂(Double-stand break, DSB),继而通过同源直接修复(Homology directed repair, HDR)[23]或非同源末端接合(Non-homologous end joining, NHEJ)[24]等方式实现对靶序列的编辑。基因编辑技术通过不同的转染技术,如脂质体、电穿孔、病毒包装、穿膜肽等[25],已经应用于疾病模型的建立[26~28]、基因治疗[29]、基因筛选[30]等各个领域。然而,基因编辑工具固有的脱靶效应[31,32]可能在实际应用中诱导非靶点编辑、错误表型或致死等副作用,如T7E1酶切试验及靶点所在片段的扩增子测序分析仅能反映局部脱靶效应,因此,只有针对全基因组的深度测序[33]才能对脱靶效应进行合理的评价。在降低脱靶效应方面,采用LNA取代[34]、2'-F、2'-O-Me、2'-O-Me 3'-硫代磷酸脂、2'-O-Me 3'-硫代磷乙酸[35~36]等化学合成碱基取代的sgRNA显著降低了Cas9脱靶效应并提高了编辑效率;替换Cas9蛋白中氨基酸序列, 可产生特异性更高或识别范围更广泛的突变体Cas9x[37]。
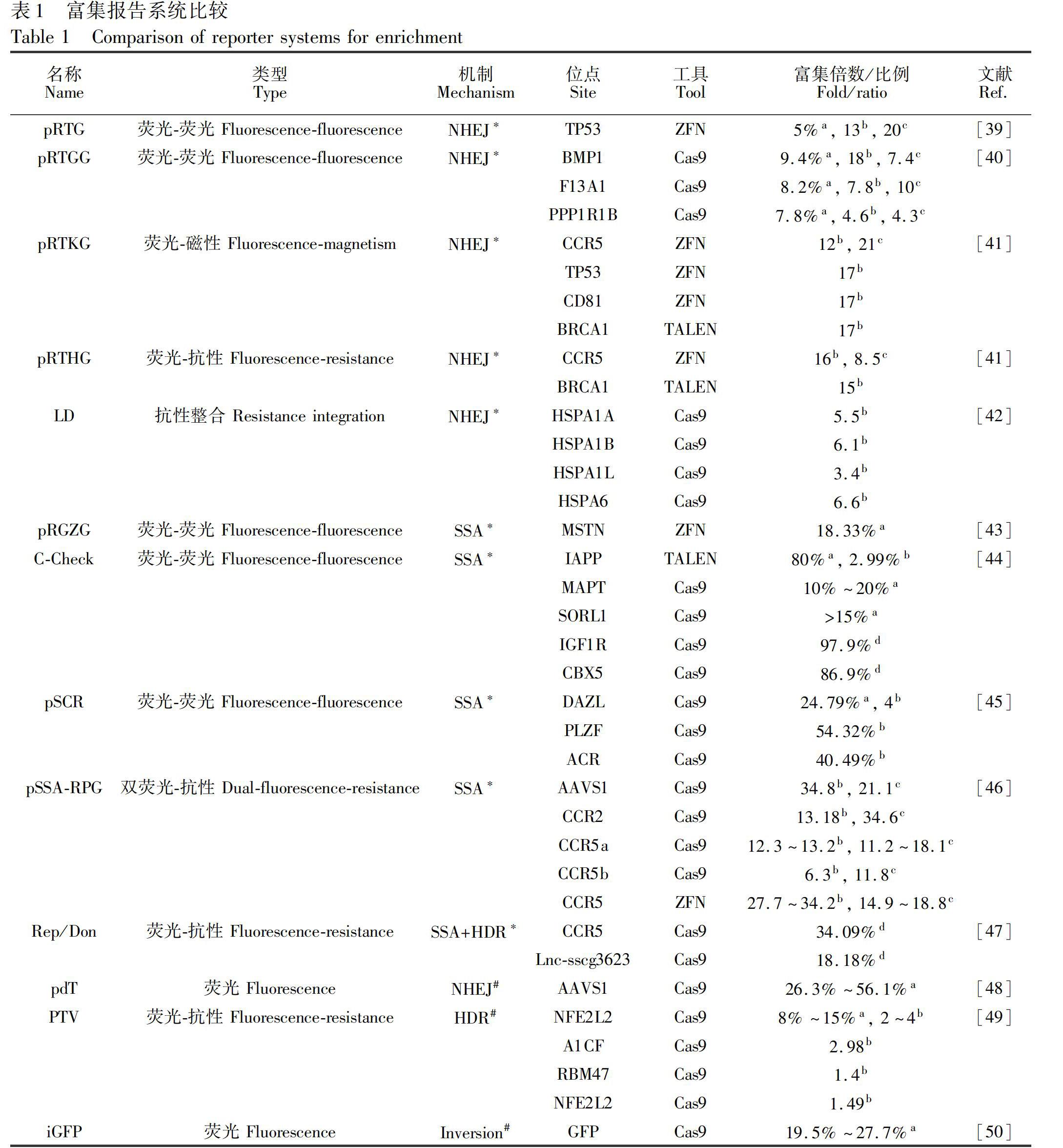
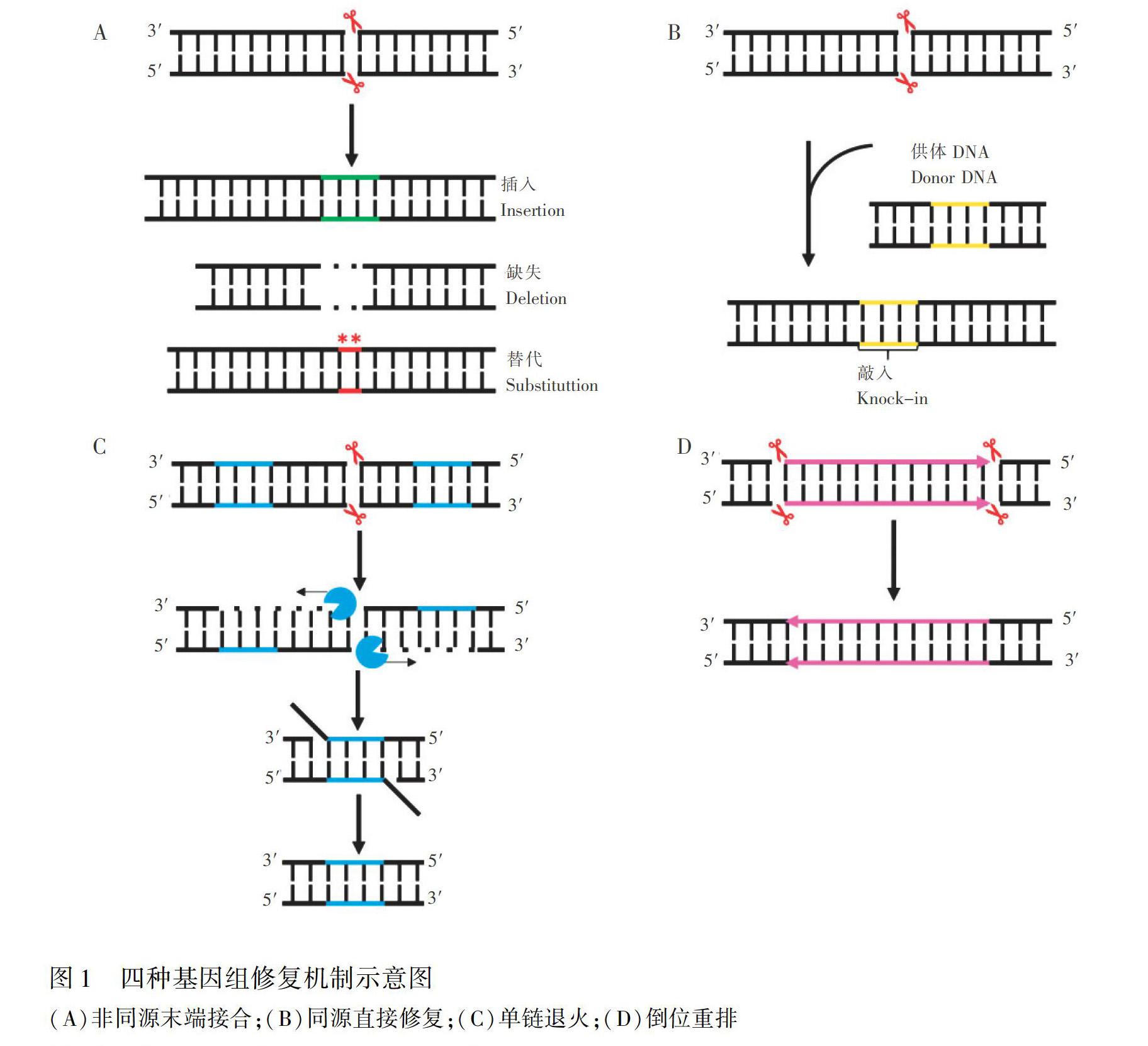
不同细胞系、基因位点和基因编辑工具等差异因素会显著影响编辑效率,导致基因编辑阳性细胞占比较少,大量野生型细胞会对突变分析产生干扰。虽然高通量深度测序能分辨突变序列,但其成本较高、数据分析难度较大,并且无法进行后续细胞培养研究。通常情况下,T7E1酶切试验可作为DNA水平编辑效率的早期评价方案用于筛选到最优的反应体系。然而,如果继续研究基因组产生突变的活细胞,则需要建立一种可直观、快速且高效富集阳性细胞的方法。目前,借助基因编辑工具酶靶向与荧光蛋白(mRFP、mCherry、DsRed、AsRED、eGFP)或抗性蛋白(嘌呤霉素、潮霉素)序列整合的外源性目的基因,再利用HDR、NHEJ、单链退火(Single strand annealing,SSA)和倒位重排[38]等修复机制诱导读码框位移来实现功能蛋白的表达,以此来表征基因编辑阳性结果的相关方法被相继报道。后续通过流式细胞荧光分选技术(Fluorescence activated cell sorting,FACS)或抗性筛选等手段完成基因编辑阳性细胞的高效富集,具有通用性强、快速检测、分析简便等优势,且此类报告系统已在多种基因编辑工具验证中得到了应用(表1)。本文主要针对基于NHEJ、SSA、HDR、倒位重排这4种基因修复机制的报告系统及其在基因编辑阳性细胞分选和富集方面的应用进行评述。
2 基因组修复机制
ZFN或TALEN通过Fok I对目的基因双链造成5'端突出的粘性末端,而Cas9依靠自身内部的RuvC和HNH两个切割域对PAM上游第3和第4个碱基之间产生平头末端[51]。 以上两种形式的DSB均會触发基因组修复。NHEJ通常导致断裂处的碱基缺失、插入或替换(图1A),这是一种有错误倾向的修复方式,一般用于基因敲除,而对病理性插入突变的两端造成两个DSB,NHEJ则会恢复基因表达[52]。HDR与SSA均属于依赖同源臂的同源重组修复机制。当含有DSB上下游同源序列的供体存在时,断裂位点依据供体序列进行精确修复,可以逆转病理突变或插入功能性基因(图1B)。SSA的触发条件是在一对较长的直接重复序列之间的间隔区发生DSB,切口处自5'端向3'端消化,随后3'突出末端处的互补序列退火完成修复(图1C)。染色体倒位重排(图1D)与多种遗传疾病和肿瘤发生相关[50],多发生于一段长序列两端同时断裂,而后该段序列方向颠倒180°。
3 细胞内游离的外源性报告系统
将靶序列整合至基于不同修复机制的外源性报告质粒,并与基因编辑工具酶表达质粒共转染至细胞中,即可通过荧光蛋白或抗性基因的表达情况来表征基因编辑的效果。
综上所述,NHEJ修复机制作为报告系统的优势在于极为简单的报告载体设计,依靠修复后产生的碱基缺失或插入使开放阅读框位移,从而实现报告功能。但是,由于缺失或插入的碱基数必须满足3n+1或3n+2(n≥0且n∈N)个,因此其它形式的突变通常无法被准确反映。
3.4 基于SSA的双荧光报告系统
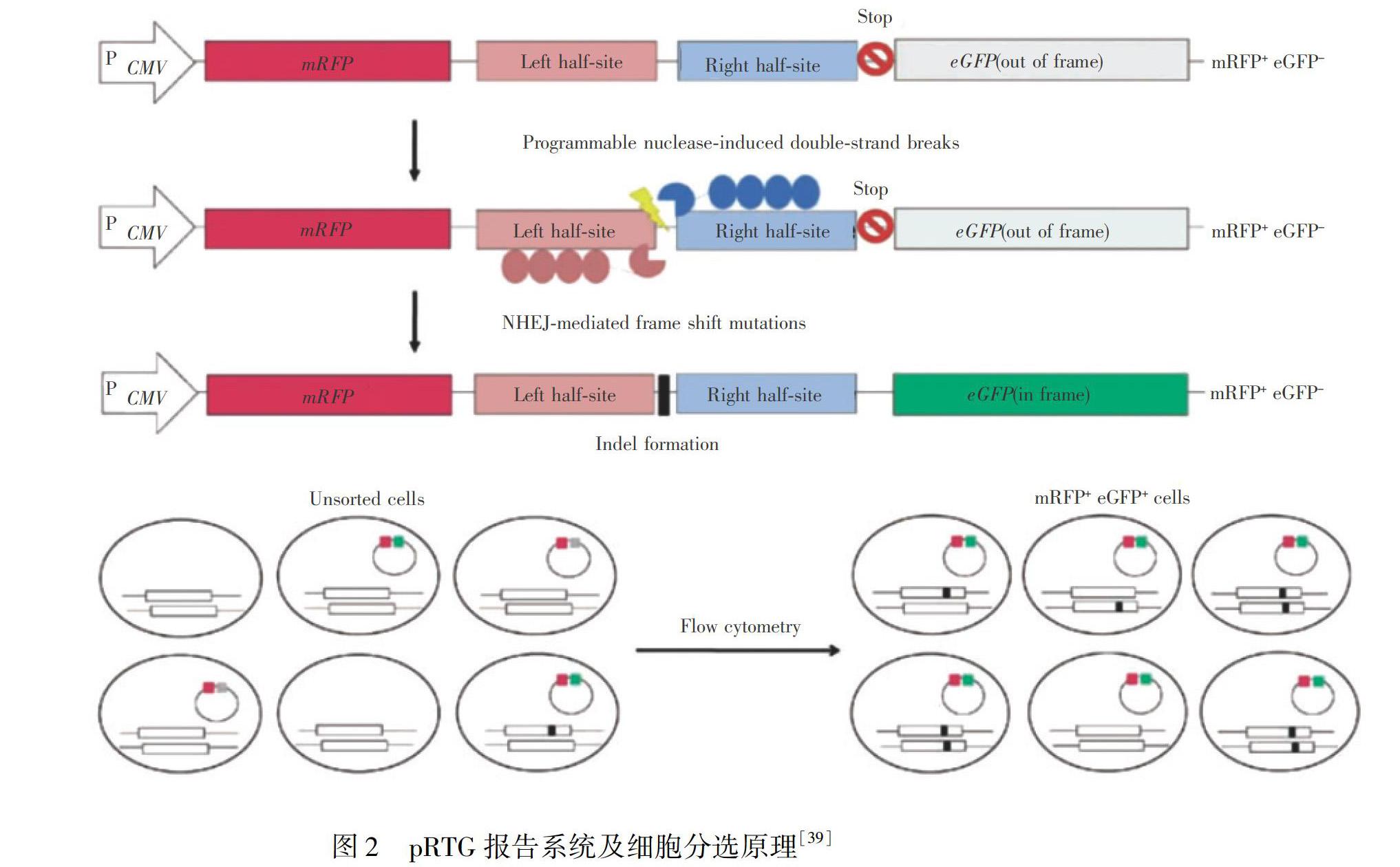
2014年, Zhang等[43]设计了一种“自降解”ZFN表达盒偶联双荧光报告系统的质粒(mRFP-GFPleft-target-ZFN-target-GFPright,pRGZG)。该系统下游eGFP序列被打断成两段(GFPleft和GFPright),二者的3'和5'端共享200 bp的直接重复序列,ZFN左右臂的表达序列插入其中,另外两对ZFN左右臂的靶标(Target)分别置于ZFN表达盒的上下游。当ZFN表达后不仅会切割基因组靶标,也会识别并切割报告质粒上的靶标并引发SSA修复,重组为完整的eGFP表达序列; 而ZFN表达盒则会被消化,使得ZFN表达终止,实现阻断ZFN持续性表达并降低细胞毒性的作用。pRGZG对ZFN诱导的MSTN突变的羊胎成纤维细胞富集到18.33%的mRFP+eGFP+细胞,且同对照组相比,细胞活性仅下降约20%,而正常转染ZFN的细胞活性下降约40%。由此可见,pRGZG系统既可高效富集基因编辑阳性细胞,又可降低ZFN的细胞毒性。
尽管ZFN的组装平台较为成熟,但每个锌指酶依靠内部3个氨基酸残基识别3个碱基,因此十分依赖上下游序列的设计,而且其细胞毒性也不容忽视[57]。与之相比,CRISPR/Cas9技术仅需要设计与靶标互补的sgRNA序列,即可配合Cas9蛋白实现对目的基因的操纵[51],极大程度地简化了实验前设计步骤。2016年,Zhou等[44]设计了基于SSA修复机制的C-Check报告质粒(图4)。C-Check由两个表达盒组成:一对用于引发SSA修复的截断eGFP表达序列和用于测定转染效率以及确立标准化的AsRED表达序列。eGFP被拆分为1~600 bp和100~720 bp两个片段,使其荧光性质被彻底干扰。同时,两部分共享500 bp的同源臂以便触发SSA修复。
为验证C-Check的功能,TALEN靶向HEK 293T细胞基因组中IAPP基因,FACS分选后AsRED+ EGFP+的细胞占比约80%,对照组则低于10%。在较难转染的原代猪成纤维细胞中,T7E1检测到299%的突变比例,Sanger测序发现約3125%的突变目的基因。Cas9/sgRNA靶向HEK 293T细胞基因组中MAPT和SORL1基因,FACS富集到117%~181%的双荧光阳性细胞,而对照组仅为237%和291%。由于C-Check报告系统可以富集到可明显区分于对照组的双阳性细胞,证明其具备较高的报告性能。研究人员将C-Check与靶向IGF1R的Cas9/sgRNA表达质粒共转染至HEK 293T细胞中,FACS依据EGFP荧光信号由弱至强分选出4组分双阳性细胞。扩增子分析显示,插入缺失的发生频率随EGFP荧光强度增强而显著提升(87%~979%),说明由CRISPR/Cas9诱导的IGF1R突变被高效富集。同样,C-Check报告系统也在癌细胞MCF-7中富集到86.9%的CBX5基因突变细胞,其mRNA水平及CBX蛋白表达量降低,进一步证实了C-Check是一种高效富集基因编辑阳性细胞的报告系统。
上述外源性报告质粒多采取核酸酶表达质粒共转染的策略,但双组分进入细胞内的比例却难以评估。CRISPR/Cas9基因编辑系统最初需要Cas9表达质粒与tracr/crRNA复合物共转染,Doudna与Charpentier将crRNA∶tracrRNA嵌合表达[58](Single-guided RNA,sgRNA),使得Cas9/sgRNA实现单一质粒表达,从而提高了转染效率。因此,若是将报告系统与Cas9/sgRNA表达质粒共包封为单一表达系统,可能会提高突变细胞富集效率。Liu等[57]将mCherry打断为两部分,各含有约300 bp的重复序列作为SSA修复臂,中间插入sgRNA靶标,再将mCherry表达盒整合到Cas9/sgRNA表达质粒中(sgRNA-mCherry-CRISPR,pSCR),copGFP用以表征转染效率。T7E1检测出copGFP+ mCherry+细胞中DAZL基因的3个靶位点的突变比例分别为24.79%、12.26%和17.18%,突变比例提高1.87、4.99和4.00倍。此外,使用双拷贝sgRNA可使鼠黑色素瘤B16细胞基因组中PLZF和ACR基因突变富集率最高达5432%和40.49%。
双荧光报告系统的优点在于一种荧光蛋白负责指示转染效率,另一种荧光蛋白指示基因编辑效率。因此,对双荧光信号阳性的细胞进行富集分析可简化和加快突变类型的研究。然而,两种荧光蛋白的发射光波长存在一定程度的重合,由此产生的非特异性背景可能干扰FACS分选,导致假双阳性细胞的混入,因此需要划定适当的阈值以便区分。
3.5 基于SSA的荧光-抗性-荧光报告系统
Ren等[46]设计了新型荧光-抗性-荧光报告质粒pSSA-RPG(DsRed-PuroR-eGFP)。其中PuroR基因被打断为上游(PuroRL)、下游(PuroRR)两部分,中间插入sgRNA识别位点以及PAM序列,PuroRL 5'末端与PuroRR 3'末端为共有的200 bp直接重复序列作为SSA修复臂,PuroRR下游以T2A剪切肽序列连接eGFP表达序列,且eGFP序列由于嘌呤霉素抗性基因序列被打断而出现在正常的ORF以外,此时细胞会呈现DsRed+eGFPPuroR。当靶标产生DSB后会引发SSA修复,使得PuroRL与PuroRR重组为完整的嘌呤霉素抗性基因,ORF复位,eGFP正常表达,细胞呈现DsRed+eGFP+PuroR+。HEK 293T细胞基因组中4个位点AAVS1、CCR2、CCR5a和CCR5b经T7E1酶切法检测,药筛组中4个位点发生突变的比例为43.9%、57.8%、27.1%和24.7%,突变倍数达34.8、13.8、12.3和6.3倍,测序结果显示突变比例分别为86.6%、72.7%、30.8%和45.0%,突变倍数达21.1、34.6、18.1和11.8倍。FACS富集DsRed+eGFP+的CCR5a突变细胞后,T7E1酶切检测到31.4%的细胞为突变阳性,突变倍数达13.2,测序显示突变比例与突变倍数分别为23.5%和11.2倍。
为了研究基于SSA修复机制的报告系统是否优于前述的NHEJ报告系统,该课题组也设计了对应的报告质粒(pNHEJ-RPG),在完整的PuroR与eGFP序列前插入终止密码子,原理同Kim等[39,40]的双荧光报告质粒一致。AAVS1位点pSSA-RPG的富集效率比pNHEJ-RPG高1.15~1.34倍,呈现出较小幅度的效率提升。然而,分析pSSA-RPG序列发现,PuroRR存在数个ATG起始密码子,或许在细胞内转录的过程中会发生下游eGFP的泄露表达,从而导致假阳性的结果出现,这是PuroR偶联eGFP双重阳性标志物共存的缺点。
近年的研究发现,细胞基因组中单一等位基因被编辑的概率明显高于双等位基因[59~61],但是,在疾病细胞模型构建、转基因动物模型构建及基因治疗等领域内,迫切需要快速筛选出双等位基因突变的细胞。Wu等[47]将pSSA-RPG改造,构建出两种整合型供体报告质粒(Reporter-intergrated donor,Rep/Don,图5),其一在PuroR-eGFP表达组件上下游插入了基因组中靶标上下游的同源臂(Rep/DonPG),另一种则将两侧同源臂内侧的报告序列更换为ZeoR-mRFP表达组件(Rep/DonZR)。将Rep/DonPG(图5A)、Rep/DonZR(图5B)与sgRNA-Cas9表达质粒共转染至细胞中,当Cas9切割基因组和Rep/Don的靶标后,Rep/Don内部发生SSA修复使抗性-荧光蛋白恢复表达,外侧同源臂则介导HDR,将PuroR-eGFP或ZeoR-mRFP整合进细胞基因组中。功能性荧光蛋白可作为初筛,反映Cas9切割活性,再进行Puro和Zeo双重药筛,即可高效筛选到双等位基因修饰的细胞。人HEK293T细胞CCR5位点和猪PK15细胞Lnc-sscg3623位点双等位基因突变比例达34.09%和18.18%,Rep/DonPG或Rep/DonZR单转染对照组的双等位基因突变率仅2.33%~6.81%,因此Rep/Don可以作为高效富集双等位基因突变的报告系统。
基于SSA修复机制的外源性报告系统,由于其较高的修复效率,且无需考虑碱基数变化,因此受到越来越多研究者的青睐。此类报告系统与基于NHEJ的报告系统有一个共同的缺陷,即荧光蛋白的泄露表达或两种荧光蛋白激发光波段的部分重合而导致的背景信号[61]。为了克服这一问题,多数研究将抗性筛选与荧光分选相结合,并利用指示性荧光蛋白进行荧光信号标准化处理,从而进一步提高特异性及富集效率。另外,Li等[56]的研究发现,提高sgRNA的表达量可直接增强Cas9切割效率,继而对报告系统的富集率起促进作用。
4 细胞基因组整合的内源性报告系统
与外源性报告系统相比,将基于相应修复机制的报告系统整合进细胞基因组中,可构建内源性报告系统,如此无需考虑多种质粒共转染的比例或转染效率等问题,而且整合后的报告系统与目的基因拷贝数一致,可更准确地反映核酸酶对靶标造成的突变,也可提高富集后的突变比例。然而,构建稳定表达细胞系常费时费力,因此整合型的内源性报告系统相关研究较少。
4.1 NHEJ介导的报告系统
D'astolfo等[48]设计了一种基于NHEJ修复机制的内源性dTomato报告系统(pdT)(图6)。pdT结构为EF1a-ATG-AAVS1-dTomato,其中AAVS1作为靶序列的同时,也使得dTomato读码框位移而无法正常表达,当Cas9对AAVS1造成切割后,下游ORF移码导致细胞呈现dTomato阳性。通过慢病毒载体构建出稳定表达的KBM7和H1人胚胎干细胞。第一轮Cas9/sgRNA转染后,KBM7细胞和H1人胚胎干细胞中dTomato强度分别达33.8%和10.2%,第二轮转染后dTomato强度升至56.1%和26.3%,同时,脱靶实验显示dTomato的阳性信号约0.2%,远低于实验组的荧光强度。上述结果表明,dTomato报告系统可高效富集突变阳性细胞,但该课题组并未对目的基因的突变情况进行分析,需要进一步的测序结果来验证该报告系统的稳定性。
4.2 HDR介导的报告系统
Piggybac转座子(PB)可实现大片段DNA的高效整合,从而构建出外源基因稳定表达的细胞系。Wen等[49]使用PB构建了供体依赖的荧光-抗性报告系统(Piggybac target vector,PTV),见图7。高活性PB转座子酶(hyPBase)将CMV-PuroR-pA-H2B-GFP-pA表达元件整合至细胞基因组,pA(polyadenylation signal)終止PuroR基因下游的GFP表达。同时,PuroR与GFP之间是一对Cas9D10A的识别序列。质粒供体含有PuroR-T2A-H2B-GFP序列,其中PuroR与H2B-GFP为PTV同源臂,PTV中的靶序列产生DSB后,PuroR-T2A-H2B-GFP序列在HDR机制下整合到报告系统。
HeLa和DLD1报告细胞系经FACS分选后,GFP+细胞分别约占8%和15%,T7E1酶切验证NFE2L2位点突变率达33.1%和9.5%,较未分选组高2~4倍。将GFP序列替换为HygroR,药筛富集到约60、120和100个存活DLD1细胞,3个基因位点(A1CF、RBM47和NFE2L2)突变率经T7E1酶切检测分别为19.7%、51.5%和29.5%,非特异性gRNA对照组则相对降低1.4~3.0倍。尽管富集后细胞基因组的靶点突变倍数提升不高,但在HDR较低发生率的前提下,此系统确实对定向插入目的基因的研究有重要意义。
4.3 基于倒位重排的报告系统
染色体中的某些区域有较强的重排倾向,可导致遗传性疾病或癌症的发生。Li等[50]构建了一种可直接反映CRISPR/Cas9诱导的序列重排的报告系统(图8)。倒置的GFP(Inverted GFP,iGFP)序列插入到CMV启动子下游,此时GFP不表达。当Cas9对GFP表达盒上下游相距约1.0 kb的位置各产生一个双链切口,iGFP的方向则可能发生倒转,使得GFP正常表达。 利用逆转录病毒将iGFP整合到鼠3T3细胞染色体中,
構建iGFP单拷贝稳定表达细胞系以实现对iGFP重排的量化。FACS分析结果表明,约23.6%±4.1%为GFP+细胞,PCR检测可见明显条带。
该iGFP重排报告系统对研究诱导染色体重排具有一定的应用前景。
现有内源性报告系统中,NHEJ相对于HDR或染色体重排而言,其修复机制更简单且发生率更高,对于阳性细胞的富集效率更高。构建稳定表达报告系统的细胞系费时费力,但却具备了外源性报告系统无法比拟的优点,如无需共转染、可实现报告系统靶标与基因组靶标拷贝数一致等。然而,由于整合的靶序列难以变更,使得每种构建出的报告细胞系仅能富集对应的目的基因,通用性较差,应用范围较窄。
5 展 望
目前,外源性游离报告系统虽然存在降解及分布不均一等问题,但其具有设计灵活、转染便捷等优势,可快速在不同细胞系中应用不同基因编辑工具进行研究;而内源性整合报告系统尽管能够更加稳定地表征基因编辑是否成功,但不可忽略细胞系构建中较长的时间成本。因此外源性报告系统的种类远多于内源性报告系统。同时,基于常见且高效的NHEJ和SSA机制以及HDR和倒位重排等低效的修复机制,衍生了荧光-荧光、荧光-抗性、荧光-磁性等报告系统。前两个报告系统的经济适用性和设计简便性更高,利用成熟的FACS技术或药筛法即可进行快速阳性富集。荧光-磁性报告系统更大的意义在于创新性,而昂贵的抗体-磁珠修饰及繁琐的洗脱步骤则降低了该系统的易用性。尽管上述报告系统实现了快速且高效的富集策略,但客观存在的内外影响因素,例如细胞系之间的异质性、不同基因位点的识别和靶向效率差异、不同基因编辑工具切割活性差异以及FACS、抗性筛选和磁性分离的灵敏度差异,均可能导致阳性细胞富集倍数或比例较低的问题。因此,为了最大程度减少或排除阴性及假阳性细胞,达到更高或完全比例的阳性细胞富集目的,可能需要两种或两种以上报告系统的共同应用或融合多种富集策略的整合报告系统进行多重筛选富集。另外,此类报告系统还需要细胞毒性试验以验证其安全性和稳定性,这将有利于确保较高的重复性以及富集后的突变细胞系的下游培养。
未来,基因编辑阳性细胞富集报告系统会不断完善并聚焦于NHEJ和SSA这两种高效修复机制。同时,应实时发展基因编辑工具,例如,重新设计报告载体的序列, 以适用于依赖转座子的非切割性CRISPR基因编辑系统,或探索其在RNA编辑或单碱基编辑中的应用价值,从而辅助并加速新型基因编辑工具的开发和拓展报告系统的新用途。此外,还应着力研究可以反映HDR这类具备临床意义的非错误性基因修复的报告系统,以促进精准基因编辑的发展和应用。
References
1 Jackson D A, Symons R H, Berg P. Proc. Natl. Acad. Sci. USA, 1972, 69(10): 2904-2909
2 Capecchi M R. Science, 1989, 244(4910): 1288-1292
3 Moore J K, Haber J E. Mol. Cell. Biol., 1996, 16(5): 2164-2173
4 Thierry A, Dujon B. Nucleic Acids Res., 1992, 20(21): 5625-5631
5 Thierry A, Perrin A, Boyer J, Fairhead C, Dujon B, Frey B, Schmitz G. Nucleic Acids Res., 1991, 19(1): 189-190
6 Smith J, Grizot S, Arnould S, Duclert A, Epinat J C, Chames P, Prieto J, Redondo P, Blanco F J, Bravo J, Montoya G, Paques F, Duchateau P. Nucleic Acids Res., 2006, 34(22): e149
7 Boissel S, Jarjour J, Astrakhan A, Adey A, Gouble A, Duchateau P, Shendure J, Stoddard B L, Certo M T, Baker D, Scharenberg A M. Nucleic Acids Res., 2014, 42(4): 2591-2601
8 Kim Y G, Cha J, Chandrasegaran S. Proc. Natl. Acad. Sci. USA, 1996, 93(3): 1156-1160
9 Wolfe S A, Nekludova L, Pabo C O. Annu. Rev. Biophys. Biomol. Struct., 2000, 29: 183-212
10 Bibikova M, Beumer K, Trautman J K, Carroll D. Science, 2003, 300(5620): 764
11 Urnov F D, Miller J C, Lee Y L, Beausejour C M, Rock J M, Augustus S, Jamieson A C, Porteus M H, Gregory P D, Holmes M C. Nature, 2005, 435(7042): 646-651
12 Miller J C, Holmes M C, Wang J, Guschin D Y, Lee Y L, Rupniewski I, Beausejour C M, Waite A J, Wang N S, Kim K A, Gregory P D, Pabo C O, Rebar E J. Nat. Biotechnol., 2007, 25(7): 778-785
13 Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, Lahaye T, Nickstadt A, Bonas U. Science, 2009, 326(5959): 1509-1512
14 Moscou M J, Bogdanove A J. Science, 2009, 326(5959): 1501
15 Mahfouz M M, Li L, Shamimuzzaman M, Wibowo A, Fang X, Zhu J K. Proc. Natl. Acad. Sci. USA, 2011, 108(6): 2623-2628
16 Christian M, Cermak T, Doyle E L, Schmidt C, Zhang F, Hummel A, Bogdanove A J, Voytas D F. Genetics, 2010, 186(2): 757-761
17 Li T, Huang S, Jiang W Z, Wright D, Spalding M H, Weeks D P, Yang B. Nucleic Acids Res., 2011, 39(1): 359-372
18 Xu S, Cao S, Zou B, Yue Y, Gu C, Chen X, Wang P, Dong X, Xiang Z, Li K, Zhu M, Zhao Q, Zhou G. Genome. Biol., 2016, 17(1): 186
19 Bolotin A, Quinquis B, Sorokin A, Ehrlich S D. Microbiology, 2005, 151(8): 2551-2561
20 Garneau J E, Dupuis M E, Villion M, Romero D A, Barrangou R, Boyaval P, Fremaux C, Horvath P, Magadan A H, Moineau S. Nature, 2010, 468(7320): 67-71
21 Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero D A, Horvath P. Science, 2007, 315(5819): 1709-1712
22 Mali P, Yang L, Esvelt K M, Aach J, Guell M, DiCarlo J E, Norville J E, Church G M. Science, 2013, 339(6121): 823-826
23 San Filippo J, Sung P, Klein H. Annu. Rev. Biochem., 2008, 77: 229-257
24 Lieber M R. Annu. Rev. Biochem., 2010, 79: 181-211
25 Aubrey B J, Kelly G L, Kueh A J, Brennan M S, O'Connor L, Milla L, Wilcox S, Tai L, Strasser A, Herold M J. Cell Rep., 2015, 10(8): 1422-1432
26 El Refaey M, Xu L, Gao Y, Canan B D, Adesanya T M A, Warner S C, Akagi K, Symer D E, Mohler P J, Ma J, Janssen P M L, Han R. Circ. Res., 2017, 121(8): 923-929
27 Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Clin. Cancer Res., 2017, 23(9): 2255-2266
28 Kurochkin I V, Guarnera E, Berezovsky I N. Trends Pharmacol. Sci., 2018, 39(1): 49-58
29 Doench J G. Nat. Rev. Genet., 2018, 19(2): 67-80
30 YUE Hua-Hua, BAO Yi-Juan, ZHOU Xiao-Ming. Acta Laser Biol. Sin., 2017, 26(6): 490-494
49 Wen Y, Liao G, Pritchard T, Zhao T T, Connelly J P, Pruett-Miller S M, Blanc V, Davidson N O, Madison B B. J. Biol. Chem., 2017, 292(15): 6148-6162
50 Li Y, Park A I, Mou H, Colpan C, Bizhanova A, Akama-Garren E, Joshi N, Hendrickson E A, Feldser D, Yin H, Anderson D G, Jacks T, Weng Z, Xue W. Genome. Biol., 2015, 16: 111
51 Jiang F, Doudna J A. Annu. Rev. Biophys., 2017, 46: 505-529
52 Cox D B, Platt R J, Zhang F. Nat. Med., 2015, 21(2): 121-131
53 Gopalappa R, Suresh B, Ramakrishna S, Kim H H. Nucleic Acids Res., 2018, 46(12): e71
54 WANG Min, SHI Huan, HUANG Xiang, LIU Xiao-Feng, QIN Yu-Feng, LIU Xiao-Hong, CHEN Yao-Sheng, HE Zu-Yong. Hereditas (Beijing), 2017, 39(1): 48-55
王 敏, 石 鹮, 黃 翔, 刘小凤, 覃玉凤, 刘小红, 陈瑶生, 何祖勇. 遗传, 2017, 39(1): 48-55
55 GAO Xiao-Tong, FU Wei, CHEN Juan-Juan, HU Bin, LI Dan-Hui, HUANG Si-Yong, LI Guo-Hui, ZHANG Yang-Ping, LIU Li, LIANG Ying-Ming. Prog. Mod. Biomed., 2016, 16(4): 652-656
高晓彤, 付 伟, 陈娟娟, 胡 彬, 李丹慧, 黄斯勇, 李国辉, 张阳萍, 刘 利, 梁英民. 现代生物医学进展, 2016, 16(4): 652-656
56 Lackner D H, Carre A, Guzzardo P M, Banning C, Mangena R, Henley T, Oberndorfer S, Gapp B V, Nijman S M, Brummelkamp T R, Burckstummer T. Nat. Commun., 2015, 6: 10237
57 Li H L, Nakano T, Hotta A. Dev. Growth Differ., 2014, 56(1): 63-77
58 Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna J A, Charpentier E. Science, 2012, 337(6096): 816-821
59 Wang Y, Li J, Xiang J, Wen B, Mu H, Zhang W, Han J. Protein Cell, 2016, 7(2): 152-156
60 He X, Tan C, Wang F, Wang Y, Zhou R, Cui D, You W, Zhao H, Ren J, Feng B. Nucleic Acids Res., 2016, 44(9): e85
61 Ye L, Wang J, Beyer A I, Teque F, Cradick T J, Qi Z, Chang J C, Bao G, Muench M O, Yu J, Levy J A, Kan Y W. Proc. Natl. Acad Sci. USA, 2014, 111(26): 9591-9596
Abstract Genome editing has become a vital tool in medical biology research. The critical mission of facilitating the progress of genome editing is to enrich positive edited cells quickly and effectively. In recent years, researchers have established various reporter systems for selection and enrichment of editing-induced positive cells based on genome repair mechanisms, such as non-homologous end joining, homology directed repair, single strand annealing and inversion, and the principle of the expression of fluorescent protein or resistance tag after genome repair. The T7E1 assay or sequencing method can analyze the mutation of enriched cells with lower background signals and higher mutation ratio. Therefore, these reporter systems can profit the characterization of genome editing effectiveness. Besides, positive cells can be cultured continuously, so this technology possesses a promising prospect of mutated cell line construction and the research of mutated cell functions. This article summarized the design principles and applications of these reporter systems and provided a reference to construct a more perfect evaluating system for genome editing.
Keywords Genome editing; Repair mechanism; Cell enrichment; Reporter system; Review
