产瓦伦西亚烯酿酒酵母的表达载体适配及发酵碳氮源优化
2020-01-14陈和锋朱晁谊李爽
陈和锋 朱晁谊 李爽
(华南理工大学生物科学与工程学院,广州 510006)
瓦伦西亚烯(Valencene)是一种天然倍半萜化合物,瓦伦西亚烯又称巴伦西亚橘烯,是一种具有似柑橘香气的黄色液体[1]。瓦伦西亚烯常见于各类柑橘果实的精油中,被广泛应用于香水、香皂等工业制造上[2],而且许多倍半萜类化工品如高附加值化合物圆柚酮(Nootkatone)也是通过瓦伦西亚烯氧化合成。除此之外,由于它具有水果香味,在商业上可被用作饮料和食品的添加剂,市场的需求量每年可达10000 kg,另外每年还有5000 kg 瓦伦西亚烯用于合成圆柚酮[3],因而应用前景巨大。
目前工业上获得瓦伦西亚烯的方法主要是以柑橘精油为原料,通过蒸馏、萃取等工艺分离出来,但是由于柑橘果实中瓦伦西亚烯的浓度较低(按重量计为0.2%-0.6%),从柑橘精油中分离获得瓦伦西亚烯的过程较为繁琐且费用较高[4]。此外,由于柑橘精油是通过将柑橘榨汁后余下的柑橘皮冷榨而获得的,而我国柑橘一般是以鲜果消费为主,柑橘榨汁工业并不发达,故也缺少它的副产物——柑橘精油,这便使得瓦伦西亚烯十分稀缺,价格昂贵,从而使瓦伦西亚烯的应用受到了极大的限制。
为了满足瓦伦西亚烯愈发增长的需求,利用基因改造优化过的微生物细胞生产瓦伦西亚烯成为经济可行的方法,因为微生物细胞具有生产周期短、培养成本低廉且发酵产物易分离等优点,可以避免在植物萃取过程中带来的高能耗、低产量及环境污染问题。而真菌中的模式生物酿酒酵母(Saccharomyces cerevisiae)则是目前最常用的基因表达宿主之一,且因为酿酒酵母是第一个完成基因组测序的真核生物,易于进行基因敲除,遗传性状也较为稳定[5-6]。因而我们需要研究萜类化合物在酿酒酵母体内的合成途径,并寻找合适的载体进行构建。为了赋予酵母倍半萜合成能力,需要人为引入相应的外源倍半萜合成酶,这些外源合成酶大部分来源于芳香植物。
MVA 途径是酿酒酵母体内合成各类萜类化合物的代谢途径,从乙酰辅酶acetyl-coA 开始,经过一系列酶作用到达中间产物FPP,并经由不同的萜类合成酶生成相应的萜类化合物,如在角鲨烯合成酶(由erg9编码)的作用下流向麦角固醇(Ergosterol)合成途径,因而为了提高通往合成目的产物的FPP流量,可以减弱erg9基因的表达。但由于麦角固醇对酵母细胞生长至关重要,无法将erg9基因的完全敲除,已有研究通过将erg9基因原有的启动子替换为较弱的启动子MET3,减少麦角固醇途径的流量,从而实现目的产物产量的提高[7-8]。此外,Özaydin等[9]发现将rox1基因敲除后,MVA 途径中间产物甲羟戊酸的表达水平得到大幅提高,其他研究也发现rox1基因在甲羟戊酸途径和麦角固醇生物合成中是抑制基因表达的转录调节器,被敲除之后可提高MVA 途径通量[10-11]。
综上,本研究尝试引入来自植物黄扁柏的瓦伦西亚烯合成酶基因(CnVS)到酿酒酵母体内,使得酿酒酵母能利用简单培养基合成瓦伦西亚烯,并结合代谢途径改造和表达载体适配提高瓦伦西亚烯产量。
1 材料与方法
1.1 材料
1.1.1 质粒及菌株 本实验中所用质粒、菌株如表1 和表2 所示。

表1 本实验所用质粒
1.1.2 主要试剂和仪器 本实验所用限制性内切酶及2×DreamTaq Green PCR 购自上海Thermo Fisher Scientific 公司,T4 连接酶购自北京New England BioLabs 公 司,2×PrimerSTAR Max Premix 及DNA Marker 购 自 大 连 的TaKaRa 公 司,ClonExpress II 重组试剂盒购自南京的Vazyme 公司,Cycle-Pure Kit 购 自OMEGA bio-tek 公 司,S.c.EasyComp Transformation Kit 购自美国的Invitrogen 公司。
本实验中所用主要仪器包括双层全温振荡摇床,PCR 基因扩增仪,美国Aligent 公司的气相色谱仪GC 7890A 及气相色谱-质谱联用仪7890-5975C,美国Waters 公司的高效液相色谱仪,上海Thermo Fisher Scientific 公司的超低温冰箱。
1.1.3 主要培养基
1.1.3.1 Luria-Bertani(LB)培养基(L-1) 称取10 g 胰蛋白胨,5 g 酵母提取物,10 g NaCl 溶于蒸馏水。配制固体培养基时,在LB 培养基中再加入20 g 琼脂粉即可。配制LB/Amp+抗性培养基时,在LB 培养基灭菌后冷却至60℃左右后,在超净工作台中以1∶1000 比例加入氨苄青霉素(100 mg/mL)即可。
1.1.3.2 三缺型SD 或SG 培养基(L-1) 配制ΔLeu-ΔTrp-ΔUra 的三缺型SD 或SG 培养基时,称取20 g葡萄糖或半乳糖溶于蒸馏水。灭菌冷却后,在超净工作台中按1∶10 的比例加入10×酵母氮源(YNB)溶液和10×DO Supplement 溶液。4℃保存。配制含某种氨基酸的单缺或双缺培养基时,在三缺型SD培养基中按1∶100 的比例加入100×的相应氨基酸母液即可。配制固体培养基时,在SD 培养基中再加入20 g 的琼脂粉即可。

表2 本实验所用菌株
1.2 方法
1.2.1CnVS基因的获取及扩增 通过查阅文献,本研究选取了来源于黄扁柏的瓦伦西亚烯合成酶CnVS(Valencene synthase fromCallitropsis nootkatensis) 进行异源表达,并从NCBI 基因库上下载获得CnVS基因序列(JX040471),将序列交由生工生物工程(上海)有限公司经密码子优化后合成。以含有CnVS基因的合成质粒pUC57-VS 为模板,用引物对VS-F、VS-R 扩增出CnVS基因片段,在片段两端引入酶切位点SacI、BamH I、XbaI 和SmaI。将扩增后得到的基因片段用1%琼脂糖凝胶电泳进行大小检测,然后再用PCR 产物纯化试剂盒Cycle-Pure Kit 纯化基因片段,用微量分光光度计检测基因片段的浓度。实验所需引物如表3 所示。
1.2.2 不同CnVS表达载体的构建 基于课题组前期构建的表达载体YEplac181-PTDH3-TADH1和YEp352-PTDH3-TADH1,用BamH I 和SmaI 分别双酶切两个载体及PCR 扩增得到的CnVS基因片段,经过琼脂糖凝胶电泳检测及试剂盒纯化后,按照基因片段∶载体片段=6∶1(n∶n)的比例配制连接体系,两个DNA 片段总质量不可以超过150 ng,反应总体系为10 μL。16℃过夜连接后,利用化学法转化入E. coliDH5α 感受态细胞中,经过colony PCR 验证后,成功得到表达载体YEplac181-PTDH3-VS-TADH1、YEp352-PTDH3-VS-TADH1。
基于课题组前期构建的表达载体YEplac181-TADH1-PTDH3-PTEF1-TCYC1,分 别 以cas1-F/181-bone-R 和181-bone-2-F/181-bone-2-R 为引物对,进行片段PCR扩增并得到两条片段backbone-1 和backbone-2,同时以质粒pUC57-VS 为模板,用引物对ADH1-VSF/TDH3-VS-R 和TEF1-VS-F/CYC1-VS-R 进行PCR 扩增,得到两条片段VS-1 和VS-2。经过上述PCR 反应 共 得 到4 个DNA 片 段backbone-1、backbone-2、VS-1 和VS-2, 利用多片段同源重组试剂盒ClonExpress MultiS,将4 个片段连接在一起,并转入E. coliDH5α 感受态细胞中,最终成功获得双向表达载体YEplac181-TADH1-VS-PTDH3-PTEF1-VS-TCYC1,用同样的方法获得双向表达载体YE352-TADH1-VS-PTDH3-PTEF1-VS-TCYC1,构建示意图如图1 所示。
1.2.3 基因敲除菌株的构建 首先从NCBI 的基因数 据 库(https://www.ncbi.nlm.nih.gov/gene/)中 下载获得S.cerevisiaeBJ5464 基因组上rox1基因的外显子序列和erg9启动子序列。然后利用CRISPy 工具(http://staff.biosustain.dtu.dk/laeb/crispy_cenpk/),获得上面2 个目的基因的所有潜在靶序列。根据CRISPy 的结果排序,选择不会与其他基因位点产生特异性匹配的序列作为gRNA 的靶序列。

表3 本实验所用引物
gRNA 表达载体是基于购自Addgene 公司的p426-PSNR52-gRNA.CAN1.Y-TSUP4(简称P426)进行构建,采取同源重组的方法。首先根据质粒P426 的序列信息,选取质粒上的一段序列作为构建2 个gRNA表达载体时的通用引物(Tong-F/R),然后以质粒上原有的gRNA 靶序列前后的序列作为基础设计上下游引物,并将rox1和erg9基因靶序列分别添加到上下游引物的5′端,形成20 bp 的同源臂,通过同源臂重组作用获得2 个单基因敲除的gRNA 表达质粒P426-rox1、P426-erg9(图2)。

图1 双向表达载体的构建示意图
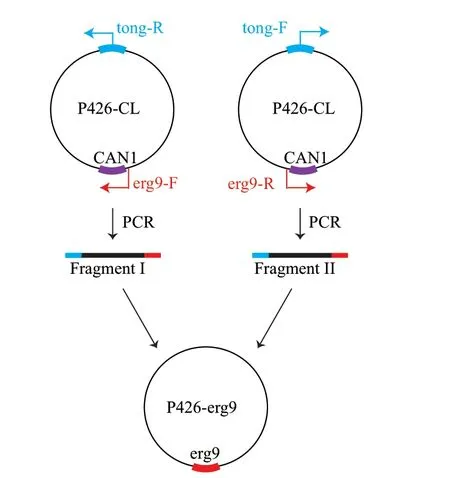
图2 gRNA 表达质粒示意图
利 用 酵 母 转 化 试 剂 盒S.c.EasyComp Transformation Kit,将Cas9 蛋白表达质粒(p414-PTEF1-Cas9-TCYC1,简称p414-Cas9)转化到S.cerevisiaeBJ5464 的感受态细胞中,最终得到可表达Cas9 蛋白的酵母菌株BJVC。随后制备酵母菌株BJVC 的感受态细胞,将500 ng gRNA 表达质粒和500 ng 对应的用于DNA 修复的同源DNA 模板共同转化到该可表达Cas9 蛋白的感受态细胞中,重组酵母细胞放在营养缺陷型平板SD/ΔUra-ΔTrp 中生长,30℃培养2-4 d。经过酵母菌落PCR 及基因测序验证,最终成功得到基因敲除菌株。
1.2.4 重组菌株双相摇瓶发酵 挑取上述构建成功的重组菌株,在对应营养缺陷型的SD 液体培养基中30℃,220 r/min 培养36-72 h,直至OD600值为1-3,然后取适量菌液加到50 mL 锥形瓶中,并在锥形瓶中加入10 mL 相应营养缺陷型的SD 液体培养基,使得锥形瓶中菌液起始OD600为0.05,加入2 mL 正十二烷覆盖在菌液上方,用于萃取瓦伦西亚烯。发酵在30℃,220 r/min 环境下培养48 h。其中原始菌株作为对照组,各基因敲除菌株作为实验组。对照组与实验组均设置3 个平行样。
探究不同碳源浓度时,设置3 个浓度梯度:20 g/L、80 g/L、140 g/L。探究不同氮源浓度时,设置了3 个浓度梯度N0、N1、N2,并设置一组将有机氮源替换成无机氮源的实验,如表4 所示。
1.2.5 瓦伦西亚烯的鉴定及产量检测 气相样品制备:发酵48 h 结束后,取出50 mL 锥形瓶,静置数分钟直至有机相与水相分离开,用移液枪吸取上层的有机相至2 mL 离心管中,14000 r/min 离心5 min。然后取500 μL 上层有机相至1.5 mL 离心管中,并加入500 μL 乙酸乙酯和2 μL 异长叶烯(作为内参,终浓度为50 μmol/L),再加入适量无水硫酸钠(吸水剂),充分振荡混匀后,14000 r/min 离心5 min。用注射器吸取离心管的上层有机相,经0.22 μm 无菌滤头过滤至气相色谱瓶中,-20℃冻存,作为GC检测的样品。
瓦伦西亚烯标样制备:为制作瓦伦西亚烯标准曲线,制备浓度梯度为10、25、50、75、100/(mmol/L)的瓦伦西亚烯标准溶液,制备方法同上。
GC-FID 检测:用气相色谱仪7890A 对制备的样品及标样进行检测,气相色谱柱为Agilent HP-5(30 m×0.32 mm;0.25 μm),使用氮气作为载气。检测方法为100℃保持10 min,然后以10℃/min 的速度匀速升温至200℃并保持8 min,结束方法。
1.2.6 发酵液水相产物的鉴定及含量检测 液相样品制备:每个取样时间点,从摇瓶中取出适量下层发酵液,14000 r/min 离心5 min,取950 μL 上清到1.5 mL 离心管中,并加入950 μL 超纯水和100 μL 10%烯硫酸,充分振荡混匀后,用0.22 μm 无菌滤头过滤至液相色谱瓶中,-20℃冻存,作为HPLC 检测的样品。
标样制备:为制作葡萄糖和乙醇的标准曲线,分别制备浓度梯度为2、4、6、8、10 g/L 的葡萄糖标准溶液,和1、2、3、4、5 g/L 的乙醇标准溶液,制备方法同上。
HPLC 检测:本研究使用高效液相色谱仪2695-2414 对制备的样品及标样进行检测。液相色谱柱为AMINEX HPX-87H(300 mm×7.8 mm),使用2.5 mmol/L 的稀硫酸作为流动相。跑样程序保持60℃,流动相速率为0.6 mL/min,分析时间为25 min。

表4 氮源浓度梯度
2 结果
2.1 双CnVS表达盒的酵母基因组整合
为构建产瓦伦西亚烯酿酒酵母菌株,利用CRISPR/Cas9 技术将双CnVS表达盒(TADH1-VS-PTDH3-PTEF1-VS-TCYC1)整合到酵母基因组的rox1基因位点上,获得该菌株BJV00 后,选取10 个单菌落进行酵母菌落PCR,扩增靶基因片段进行测序,验证双CnVS表达盒整合效果。图3 为双CnVS表达盒整合基因组的菌落PCR 结果,从图中看到3 号单菌落在rox1位点可扩增出两个CnVS表达盒的片段,基因测序结果也正确,说明成功将双CnVS表达盒整合到酵母基因组上。
图3 双CnVS 表达盒整合的酵母菌落PCR 结果
2.2 瓦伦西亚烯产量与细胞生长曲线
成功实现rox1基因的敲除并将双CnVS表达盒整合到rox1位点后,获得可合成瓦伦西亚烯的出发菌株BJV00,挑取该菌株的单克隆接种进行双相摇瓶发酵,探究瓦伦西亚烯产量变化与细胞生长曲线的关联,结果如图4 所示。从图中可看出,瓦伦西亚烯的增长曲线与细胞生长曲线基本保持一致,最高达到4.16 mg/L,而在14 h 葡萄糖被消耗完后细胞增长开始变缓,同时瓦伦西亚烯的增长也变缓,说明瓦伦西亚烯的合成是生长偶联型。从乙醇曲线和甘油曲线可以发现在葡萄糖未消耗完前,两者均是处于不断积累的状态,当葡萄糖消耗完后,两者含量开始下降,说明细胞开始消耗乙醇及甘油进行生长和瓦伦西亚烯合成。
2.3 erg9基因下调对瓦伦西亚烯产量的影响
在BJV00 菌株基础上,我们引入对erg9基因的下调,在获得基因敲除酵母菌株后,同样选取10 个单菌落进行酵母菌落PCR,扩增靶基因片段进行测序,验证各基因敲除效果。图5 为基因敲除↓erg9的酵母菌落PCR 结果,从图中可看到各单菌落均扩增出了条带单一的目的条带,挑取1-3 对应的PCR产物直接送测,测序结果显示均为阳性,即成功对靶位点进行了定点修饰,获得菌株BJV01,基因测序结果如图6 所示。

图4 瓦伦西亚烯与细胞生长曲线

图5 转化子酵母菌落PCR 结果
挑取菌株BJV01 单菌落接种进行摇瓶发酵,发酵结果如图7 所示,对erg9基因下调后,菌体生长与原始菌株保持一致,并未有明显影响,而产量比原始菌株提高了约1.7 倍,达到了6.63 mg/L,表明erg9基因的下调对瓦伦西亚烯产量起到促进作用。
2.4 不同碳氮源浓度对瓦伦西亚稀产量的影响

图6 基因敲除的测序结果图
基于BJV01 菌株,对培养基的碳氮源浓度进行探究。碳源浓度选取了20 g/L、80 g/L、140 g/L 进行摇瓶发酵,结果如图8 所示,初始糖浓度越高,菌体生长量也越高,80 g/L 糖浓度的产量最高,达到8.1 mg/L,提高了约1.3 倍,而在此细胞产量上,20 g/L糖浓度与80 g/L 糖浓度一致,高于140 g/L 糖浓度。氮源浓度设置参考1.2.4,发酵结果如图9 所示,提高氮源浓度同样可以提高菌体生长量,最高的N2 组达到33,而S 组中将氮源里的蛋白胨替换成无机氮源硫酸铵后,菌体生长量大幅下降,仅有8.45。N1组的产量最高,达到11.74 mg/L,比N0 组提高了约1.3 倍,而在比细胞产量上却是S 组最高,远高于另外3 组。

图7 基因敲除菌株发酵结果

图8 不同碳源浓度发酵结果

图9 不同氮源浓度发酵结果
2.5 不同CnVS表达载体的构建
以YEplac181-PTDH3-VS-TADH1为例,在得到阳性克隆转化子后,挑取单菌落接种培养,然后提取质粒进行双酶切验证。如图10 所示,重组质粒经双酶切后得到线性YEplac181 片段和CnVS基因片段,大小值分别为约6500 bp 和1800 bp 左右,均与理论值相符,表明成功构建重组质粒YEplac181-PTDH3-VS-TADH1,同理验证重组表达载体YEplac352-PTDH3-VS-TADH1,测序结果正确,均构建成功。
双向表达载体以YEplac181-TADH1-VS-PTDH3-PTEF1-VS-TCYC1为例,VS-1 和VS-2 基因片段是以pUC57-VS 为模板PCR 扩增得到,backbone-1 和backbone-2片段是以YEplac181-TADH1-PTDH3-PTEF1-TCYC1为模板PCR 扩增得到,图11-A 为4 个片段的琼脂糖凝胶电泳图,从结果可看出每个片段均得到单一的目的条带,大小也符合理论值。片段通过同源重组连接在一起后,挑选阳性克隆提取质粒进行酶切验证,如图11-B 所示,大小符合预期,测序结果正确。
2.6 不同表达载体对瓦伦西亚烯产量的影响
基于载体YEplac181 和YEp352 构建了不同CnVS表 达 载 体p181-VS、p352-VS、p181-2VS、p352-2VS,并导入酿酒酵母BJ5464 体内得到菌株BJV02、BJV03、BJV04、BJV05, 发 酵 结 果 如图12 所示,结果表明在只含单CnVS表达盒的情况下,BJV03 产量是BJV02 产量的2.35 倍,增加一个CnVS表达盒后,BJV04 比BJV02 提高了约2.5 倍,但BJV05 相比BJV03 并没有明显提高。且在含双CnVS表达盒时,BJV04 和BJV05 并无明显差距。为了进一步提高产量,将最优表达载体p181-2VS 转入双基因敲除菌株BJV06(↓erg9Δrox1)中,获得菌株BJV07,经过发酵最终获得17.54 mg/L 的产量,比双CnVS表达盒在酵母基因组上的BJV01 提高了2.6 倍。

图10 重组表达载体酶切验证结果

图11 双向表达载体片段PCR 扩增结果
3 讨论
本研究通过在酿酒酵母BJ5464 体内引入来自植物黄扁柏的CnVS基因,并将两个CnVS表达盒整合入酵母基因组的rox1位点上,使得酵母细胞能利用简单碳源合成瓦伦西亚烯,同时rox1位点的敲除也可以解除对MVA 途径的抑制[10-11],初始产量为4.16 mg/L。为进一步提高产量,对麦角固醇合成途径的关键基因erg9进行下调,增加通往瓦伦西亚烯途径的碳流量,发酵产量的提升证明erg9基因的下调对瓦伦西亚烯产量起到促进作用,这也与已有文献报道相符[7,12-15],也说明下调或敲除竞争途径的改造策略的有效性[16]。
探究不同碳氮源浓度对产量影响时,碳氮源浓度的提高均能提高细胞生长积累,但产量却并没有与碳氮源浓度完全成正比,可能原因是碳氮源浓度过高时,菌体的生长速率过快,代谢能量均用于细胞生长,而并未用于瓦伦西亚稀的合成,所以碳氮源浓度提高对产量的提升作用并不明显,这可能也是随着碳氮源浓度提高,比细胞产量反而下降的原因。另外也说明虽然瓦伦西亚稀的合成是生长偶联型,但并非菌体生长积累越高,瓦伦西亚稀产量越高,而是需要将细胞生长速率与产物合成速率控制在一定比值范围内,这也是后续需要进一步探究的。还有研究表明细胞体内氧化还原失衡时,会导致代谢流溢出,流向乙醇甘油等副产品的合成,造成能量的损失,不利于目的产物的积累[17-18],控制低糖浓度发酵有利于减少副产物的产生,因而发酵培养基有待进一步优化。
在不同CnVS表达载体的实验中,单CnVS表达盒的情况下,YEp352 载体的表现要优于YEplac181载体,说明不同载体也会对瓦伦西亚烯产量造成影响,可能原因一是YEp352 载体拷贝数要高于YEplac181,提高了整体的酶活,后续可以通过检测两者的拷贝数来进行验证;二是YEp352 载体上的某部分元件促进CnVS的表达,从而提高了瓦伦西亚烯产量,这仍有待继续探究。另外在同样的基因敲除背景下,CnVS表达盒在质粒上时的瓦伦西亚烯产量要远优于CnVS表达盒整合到基因组上的产量,猜测是基因组上CnVS基因拷贝数较低,导致酶活较低的原因[19-20],因而提高CnVS基因的拷贝数是进一步提高产量的关键[21-23]。

图12 含不同CnVS 表达载体菌株的发酵结果
4 结论
本研究基于酿酒酵母菌株BJ5464,引入瓦伦西亚烯合成酶,并结合代谢途径改造、培养基优化以及表达载体适配性等策略,最终获得最高产量为17.54 mg/L 的酵母工程菌株,比出发菌株提高了约4.2倍,对利用其他微生物表达高价值化合物具有重要参考意义。
