硝酸羟胺-(H2O)n复合物氢键相互作用的密度泛函研究
2019-12-06刘建国杜文革邱从礼朱晨光
刘建国, 杜文革, 邱从礼, 夏 杰, 张 倩, 朱晨光
(1. 中国人民解放军63870部队, 华阴 714200; 2. 中国人民解放军陆军工程大学, 石家庄 050003)
1 引 言
硝酸羟胺(Hydroxylamine nitrate,HAN)是羟胺的硝酸盐,它由还原组分羟胺和氧化组分硝酸共同组成[1]. 硝酸羟胺不仅可以作为还原剂用于放射性元素的提取、核原料的处理以及核废料的再生[2],也可以作为氧化剂用于炮弹的发射和火箭的推进[3]. 以硝酸羟胺为氧化剂制备的液体推进剂是一种新型绿色液体推进剂,该推进剂密度大、比冲高、冰点低、安全无毒,是液体火箭发动机的理想燃料之一[4, 5],也广泛的应用于航天领域中微、小型推进器的推力系统[6, 7]. 目前美国军方已研制出硝酸羟胺基发射药LP1846和LP1898,美国宇航局(NASA)路易斯研究中心在军方液体发射药的基础上开发出用于航天推进的HAN基单元推进剂[8].
近年来,人们围绕硝酸羟胺溶液开展了广泛的研究,Wei[9]等使用量热法评估了质量分数为24%的硝酸羟胺溶液的热分解特征,认为分解过程中存在自催化分解特征;Koh[10]等采用铜电极和铝电极对硝酸羟胺溶液在电压下的分解特性进行了研究,并分析了溶液浓度和电源功率对分解特征的影响;Rachid[11]等研究了硝酸羟胺液体推进剂的热分解和催化分解过程,并揭示了铱催化剂对硝酸羟胺液体推进剂的催化分解机理. 但目前对硝酸羟胺-(H2O)n复合物的氢键作用详细研究尚无报道,因此采用密度泛函的方法对硝酸羟胺-(H2O)n复合物的几何构型以及氢键相互作用进行研究,以期为新型硝酸羟胺基液体推进剂的研发及其优越性能的深入研究提供理论依据.
2 计算方法
文献[12]采用密度泛函B3LYP/6-311++G(d, p)方法对HAN盐进行了研究,本文根据该方法计算出的几何结构参数与文献[13]中给出的结果吻合的较好,因此采用B3LYP方法在6-311++G(d, p)基组水平上对HAN-(H2O)n复合物的可能稳定构型进行几何优化,并对各构型进行振动分析,计算结果中均无虚频,说明得到的分子结构对应于能量的极小值点. 在此基础上,对优化所得构型进行自然键轨道(natural bond orbital,NBO)分析,以揭示氢键相互作用的本质[14].
使用MP2/6-311++G(d, p)方法,经基组叠加误差(basis set superposition error,BSSE)和零点能(zero point energy,ZPE)校正对相互作用能进行计算. 计算所用公式如下[15, 16]:
ΔE=EA…B-EA-EB
其中,ΔE为相互作用能,EA…B为构型的总能量,EA和EB为单体构型的能量,全部计算在Gaussian09[17]软件包中进行.
3 结果与讨论
3.1 硝酸羟胺-(H2O)n复合物构型分析
采用密度泛函方法,在B3LYP在6-311++G(d, p)基组水平上对HAN-(H2O)n复合物所有可能构型进行几何优化,得到HAN-H2O稳定构型6个,HAN-(H2O)2稳定构型8个,相应的几何结构和主要参数分别如图1和图2所示. 使用MP2/6-311++G(d, p)方法对HAN-(H2O)n复合物总能量、零点能以及经BSSE和ZPE校正前后的相互作用能进行计算,计算结果如表1所示.
HAN-H2O复合物的6个异构体A、B、C、D、E和F均属于C1点群,其中构型B的相互作用能绝对值最大为52.821 kJ·mol-1,是HAN-H2O复合物中最稳定的构型;构型F的相互作用能绝对值最小为45.519 kJ·mol-1,是HAN-H2O复合物中稳定性最差的构型. 各构型相互作用能绝对值排列顺序为构型B>A>D>C>E>F,由此可见,HAN-H2O复合物稳定性排列顺序为构型B>A>D>C>E>F.
构型B中含有一个N…H-O氢键和两个O…H-O氢键. 其中,N…H-O氢键是由N原子作为质子受体,H原子作为质子供体所形成;O…H-O氢键是由O原子作为质子受体,H原子作为质子供体所形成. N…H-O氢键键长为0.1793 nm,键角为174.55°,两个O…H-O氢键键长分别为0.1587 nm和0.1965 nm,键角分别为174.49°和172.93°. 构型F中含有三个O…H-O氢键,均由O原子作为质子受体,H原子作为质子供体所形成. 三个O…H-O氢键键长分别为0.1626 nm、0.1762 nm和0.2131 nm,键角分别为174.38°、164.63°和139.04°.
HAN-(H2O)2复合物的8个异构体G、H、I、J、K、L、M和N也均属于C1点群,其中构型L的相互作用能绝对值最大为73.349 kJ·mol-1,是HAN-(H2O)2复合物中最稳定的构型;构型J的相互作用能绝对值最小为60.869 kJ·mol-1,是HAN-(H2O)2复合物中稳定性最差的构型. 各构型相互作用能绝对值排列顺序为构型L>K>N>M>H>G>I>J,由此可知,HAN-(H2O)2复合物稳定性排列顺序为构型L>K>N>M>H>G>I>J.
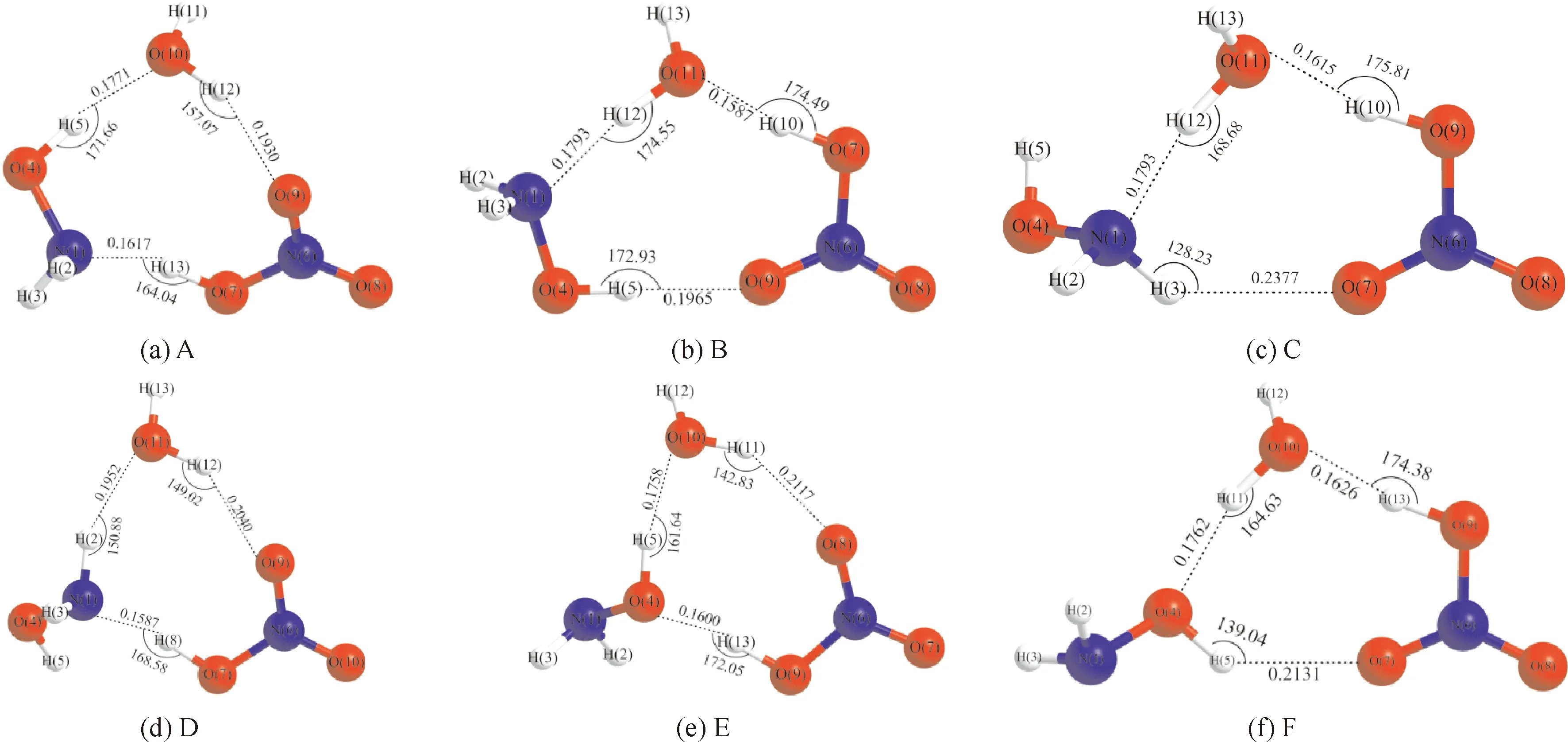
图1 HAN-H2O复合物平衡结构Fig. 1 The optimized structures of hydroxylamine nitrate-H2O complexes (bond length/nm, bond angle/°)
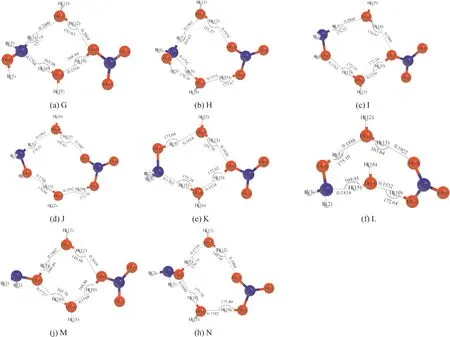
图2 HAN-(H2O)2复合物平衡结构Fig. 2 The optimized structures of hydroxylamine nitrate-(H2O)2 complexes (bond length/nm, bond angle/°)
构型L中含有一个N…H-O氢键和三个O…H-O氢键. 其中,N…H-O氢键是由N原子作为质子受体,H原子作为质子供体所形成;O…H-O氢键是由O原子作为质子受体,H原子作为质子供体所形成. N…H-O氢键的键长为0.1818 nm,键角为163.93°,O…H-O氢键的键长分别为0.1532 nm、0.1888 nm、0.1922 nm,键角分别为172.64°、175.10°和163.64°. 构型J中含有O…H-N氢键和三个O…H-O氢键,均由O原子作为质子受体,H原子作为质子供体所形成. O…H-N氢键的键长为0.1993 nm,键角为178.51°,三个O…H-O氢键的键长分别为0.1592 nm、0.1768 nm和0.1945 nm,键角分别为172.76°、170.69°和159.85°. 与N…H-O氢键键长相比,氢键O…H-N的键长更长,其氢键作用更弱,这主要是由于和O-H键相比,N-H键是弱电子受体,O原子提供的孤对电子与N-H键的相互作用较小,导致O…H-N键相对较长,氢键作用较弱.
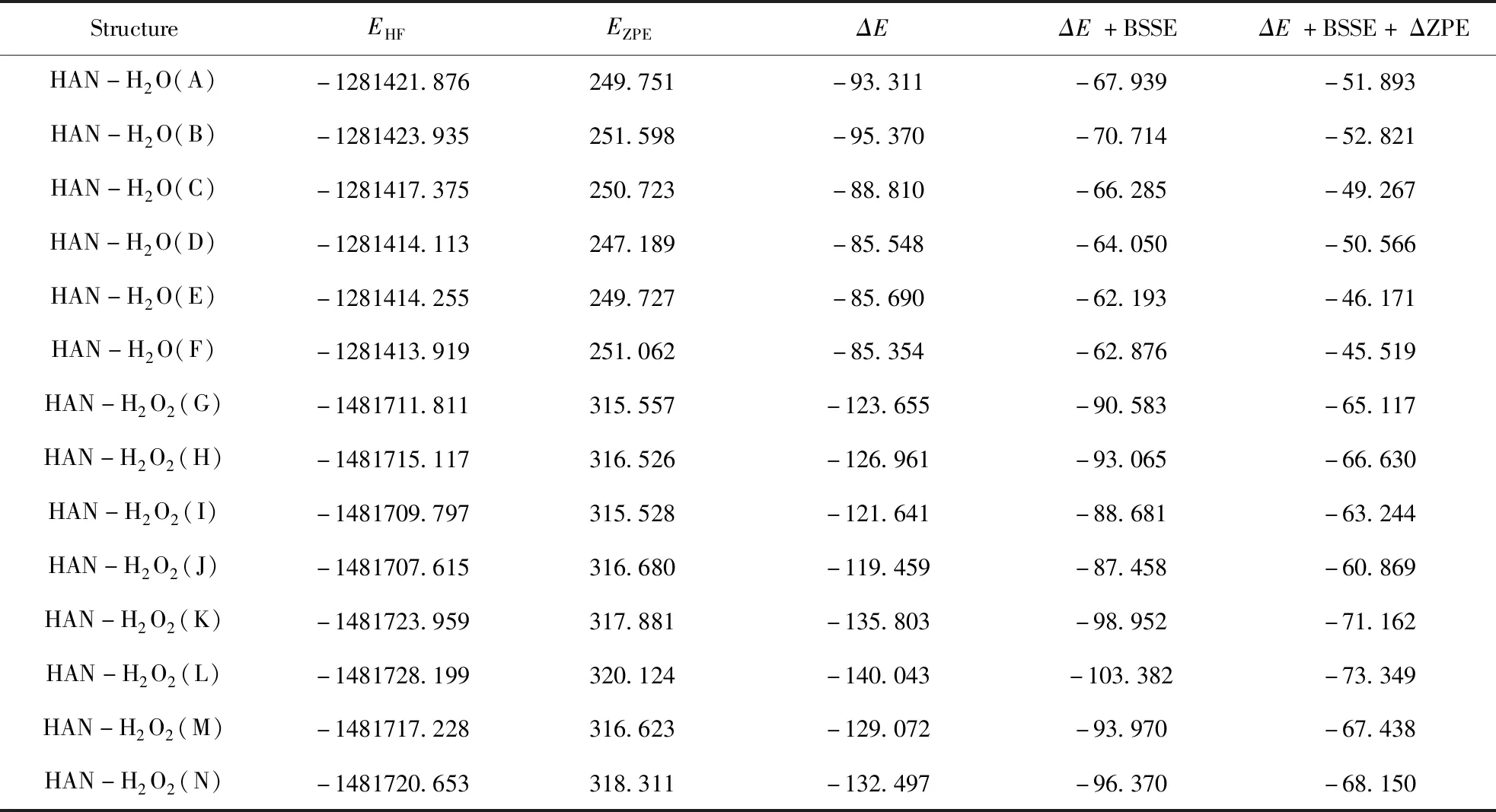
表1 MP2/6-311++G(d, p)的总能量、零点能和相互作用能
注:EHF为总能量,kJ·mol-1;EZPE为零点能,kJ·mol-1;ΔE为未校正的相互作用能,kJ·mol-1;ΔE+BSSE为经基组叠加误差校正的相互作用能,kJ·mol-1;ΔE+BSSE+ΔZPE为经基组叠加误差和零点能校正的相互作用能,kJ·mol-1.
3.2 自然键轨道(NBO)分析
在MP2/6-311++G(d, p)水平下,对HAN-(H2O)n复合物进行自然键轨道分析. 表2中列出电子供体(Donor)轨道i、电子受体(Acceptor)轨道j以及相互作用的稳定化能E(j),部分稳定化能较小未列于表中. 稳定化能E与相互作用的强度成正比,稳定化能越大,i与j的相互作用越强[18].
HAN-H2O复合物的6个异构体中构型A的N(1)孤对电子(1)对O(7)-H(13)的σ反键轨道的稳定化能为277.40 kJ·mol-1,O(10)的孤对电子(2)对O(4)-H(5)的σ反键轨道的稳定化能为77.028 kJ·mol-1;构型B的N(1)的孤对电子(1)对O(11)-H(12)的σ反键轨道的稳定化能为122.13 kJ·mol-1,O(11)的孤对电子(2)对O(7)-H(10)的σ反键轨道的稳定化能为188.28 kJ·mol-1. 由此可以得出结论,构型A和B的氢键作用主要来自于N的孤对电子与邻近的O-H反键轨道之间,O的孤对电子与邻近的O-H反键轨道之间. 类似分析可知,构型C和D的氢键作用主要来自于N的孤对电子与邻近的O-H反键轨道之间,O的孤对电子与邻近的O-H和N-H反键轨道之间. 构型E和F的氢键作用主要来自于O的孤对电子与邻近的O-H反键轨道之间. HAN-H2O复合物的6个异构体中稳定化能之和均在200 kJ·mol-1以上,形成较强氢键,HAN-H2O复合物的结构比较稳定.
HAN-(H2O)2复合物的8个异构体中构型G的N(1)孤对电子(1)对O(14)-H(16)的σ反键轨道的稳定化能为121.63 kJ·mol-1,O(14)的孤对电子(2)对O(9)-H(10)的σ反键轨道的稳定化能为217.99 kJ·mol-1;构型H的N(1)孤对电子(1)对H(3)-O(14)的σ反键轨道的稳定化能为157.74 kJ·mol-1,O(14)的孤对电子(2)对O(8)-H(15)的σ反键轨道的稳定化能为199.33 kJ·mol-1. 由此分析可知,构型G和H的氢键作用主要来自于N的孤对电子与邻近的O-H反键轨道之间,O的孤对电子与邻近的O-H轨道之间. 类似分析可以得出结论,构型I、J、M和N的氢键作用主要来自于O的孤对电子与O-H反键轨道之间;构型K和L的氢键作用主要来自于N的孤对电子与O-H反键轨道之间,O的孤对电子与O-H反键轨道之间. HAN-(H2O)2复合物的8个异构体中稳定化能之和均在250 kJ·mol-1以上,氢键作用较强,HAN-(H2O)2复合物的结构比较稳定.
表2 MP2/6-311++G(d, p)水平上复合物的NBO分析部分结果
Table 2 Parts of calculated results of hydroxylamine nitrate-(H2O)ncomplexes at MP2/6-311++G(d, p) level by NBO analysis
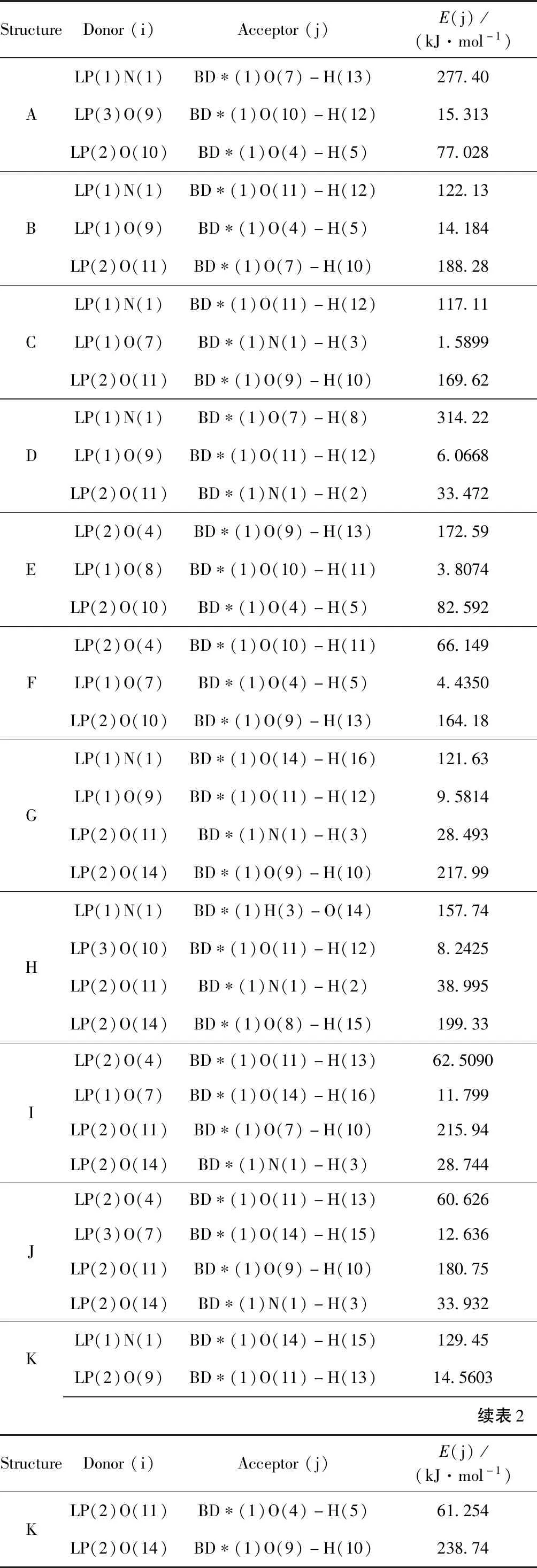
StructureDonor(i)Acceptor(j)E(j)/(kJ·mol-1)ALP(1)N(1)BD∗(1)O(7)-H(13)277.40LP(3)O(9)BD∗(1)O(10)-H(12)15.313LP(2)O(10)BD∗(1)O(4)-H(5)77.028BLP(1)N(1)BD∗(1)O(11)-H(12)122.13LP(1)O(9)BD∗(1)O(4)-H(5)14.184LP(2)O(11)BD∗(1)O(7)-H(10)188.28CLP(1)N(1)BD∗(1)O(11)-H(12)117.11LP(1)O(7)BD∗(1)N(1)-H(3)1.5899LP(2)O(11)BD∗(1)O(9)-H(10)169.62DLP(1)N(1)BD∗(1)O(7)-H(8)314.22LP(1)O(9)BD∗(1)O(11)-H(12)6.0668LP(2)O(11)BD∗(1)N(1)-H(2)33.472ELP(2)O(4)BD∗(1)O(9)-H(13)172.59LP(1)O(8)BD∗(1)O(10)-H(11)3.8074LP(2)O(10)BD∗(1)O(4)-H(5)82.592FLP(2)O(4)BD∗(1)O(10)-H(11)66.149LP(1)O(7)BD∗(1)O(4)-H(5)4.4350LP(2)O(10)BD∗(1)O(9)-H(13)164.18GLP(1)N(1)BD∗(1)O(14)-H(16)121.63LP(1)O(9)BD∗(1)O(11)-H(12)9.5814LP(2)O(11)BD∗(1)N(1)-H(3)28.493LP(2)O(14)BD∗(1)O(9)-H(10)217.99HLP(1)N(1)BD∗(1)H(3)-O(14)157.74LP(3)O(10)BD∗(1)O(11)-H(12)8.2425LP(2)O(11)BD∗(1)N(1)-H(2)38.995LP(2)O(14)BD∗(1)O(8)-H(15)199.33ILP(2)O(4)BD∗(1)O(11)-H(13)62.5090LP(1)O(7)BD∗(1)O(14)-H(16)11.799LP(2)O(11)BD∗(1)O(7)-H(10)215.94LP(2)O(14)BD∗(1)N(1)-H(3)28.744JLP(2)O(4)BD∗(1)O(11)-H(13)60.626LP(3)O(7)BD∗(1)O(14)-H(15)12.636LP(2)O(11)BD∗(1)O(9)-H(10)180.75LP(2)O(14)BD∗(1)N(1)-H(3)33.932KLP(1)N(1)BD∗(1)O(14)-H(15)129.45LP(2)O(9)BD∗(1)O(11)-H(13)14.5603续表2StructureDonor(i)Acceptor(j)E(j)/(kJ·mol-1)KLP(2)O(11)BD∗(1)O(4)-H(5)61.254LP(2)O(14)BD∗(1)O(9)-H(10)238.74
注:E为稳定化能,kJ·mol-1;BD为成键轨道;BD*为反键轨道;LP为孤对电子;对BD和BD*来讲,(1)和(2)分别为σ轨道和π轨道;对于LP来讲,(1)和(2)分别为第一和第二孤对电子.
3.3 振动光谱分析
水分子的振动频率变化是反映氢键形成的重要光谱特征,表3给出了硝酸羟胺与水形成氢键后,H2O中的O-H键长与H2O单独存在时相比键长、振动频率的变化. 从表中可以看出,当复合物形成氢键后,H2O中的O-H键长均表现出伸长,变化范围在0.000798 nm~0.004208 nm之间,振动频率明显减小,表现出明显的红移. 振动模式受到氢键作用的直接影响,硝酸羟胺-(H2O)n复合物形成的是典型的红移型氢键. 另外,通过对比表2和表3可以发现,红移增大的程度与复合物稳定化能的变化趋势基本一致,这可能是由于孤对电子与相邻反键轨道相互作用越强,导致氢键键长和频率变化更大.
表3 B3LYP/6-311++G(d, p)水平上形成氢键后水分子O-H的键长的相应的伸缩振动的变化
Table 3 The O-H stretching vibration of water in hydroxylamine nitrate-(H2O)nat B3LYP/6-311++G(d, p) level
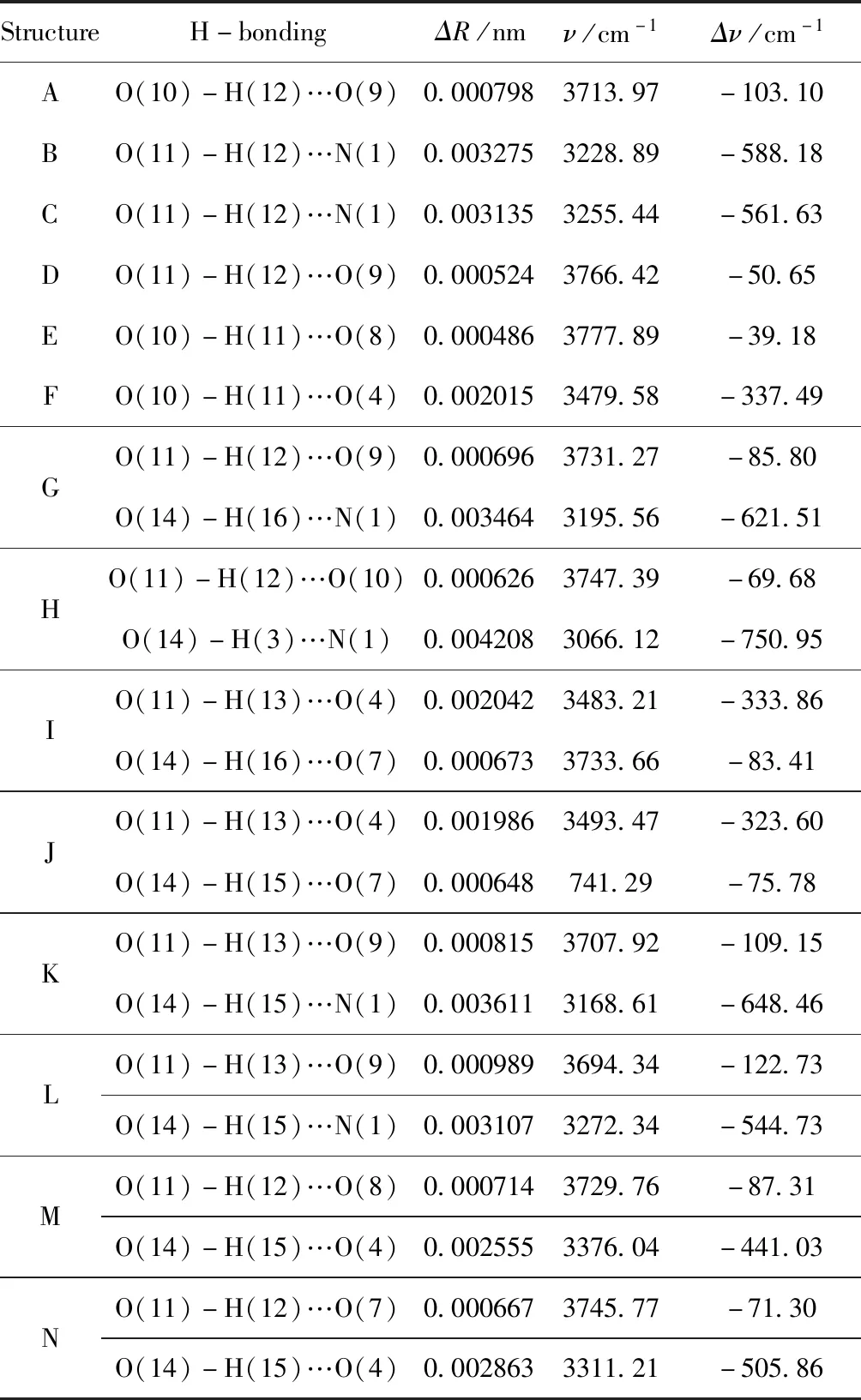
StructureH-bondingΔR/nmν/cm-1Δν/cm-1AO(10)-H(12)…O(9)0.0007983713.97-103.10BO(11)-H(12)…N(1)0.0032753228.89-588.18CO(11)-H(12)…N(1)0.0031353255.44-561.63DO(11)-H(12)…O(9)0.0005243766.42-50.65EO(10)-H(11)…O(8)0.0004863777.89-39.18FO(10)-H(11)…O(4)0.0020153479.58-337.49GO(11)-H(12)…O(9)0.0006963731.27-85.80O(14)-H(16)…N(1)0.0034643195.56-621.51HO(11)-H(12)…O(10)0.0006263747.39-69.68O(14)-H(3)…N(1)0.0042083066.12-750.95IO(11)-H(13)…O(4)0.0020423483.21-333.86O(14)-H(16)…O(7)0.0006733733.66-83.41JO(11)-H(13)…O(4)0.0019863493.47-323.60O(14)-H(15)…O(7)0.000648741.29-75.78KO(11)-H(13)…O(9)0.0008153707.92-109.15O(14)-H(15)…N(1)0.0036113168.61-648.46LO(11)-H(13)…O(9)0.0009893694.34-122.73O(14)-H(15)…N(1)0.0031073272.34-544.73MO(11)-H(12)…O(8)0.0007143729.76-87.31O(14)-H(15)…O(4)0.0025553376.04-441.03NO(11)-H(12)…O(7)0.0006673745.77-71.30O(14)-H(15)…O(4)0.0028633311.21-505.86
注:ΔR为O-H键长的变化,nm;ν为O-H键振动频率,cm-1;Δν为O-H键振动频率的变化,cm-1.
4 结 论
(1)用Gaussian09软件在B3LYP/6-311++G(d, p)基组水平上对硝酸羟胺-(H2O)n复合物的平衡结构进行优化,得到硝酸羟胺-H2O复合物稳定构型6个,硝酸羟胺-(H2O)2复合物稳定构型8个,所有构型均属于C1点群.
(2)经基组叠加误差和零点能校正计算得到硝酸羟胺-(H2O)n复合物的相互作用能,由此得出,硝酸羟胺-H2O复合物稳定性排列顺序为构型B>A>D>C>E>F,硝酸羟胺-(H2O)2复合物稳定性排列顺序为构型L>K>N>M>H>G>I>J.
(3)HAN-H2O复合物的稳定化能之和均在200 kJ·mol-1以上,HAN-(H2O)2复合物的稳定化能之和均在250 kJ·mol-1以上,酸羟胺-(H2O)n复合物复合物中存在较强的氢键作用.
(4)通过振动光谱分析,在硝酸羟胺-(H2O)n复合物中,水中H-O伸缩振动频率明显红移,且红移增大的程度与复合物稳定化能的变化趋势基本一致.
