非综合征型唇腭裂核心家庭致病基因突变筛查
2019-11-26茆武熊竹友李薇李光早
茆武,熊竹友,李薇,李光早
(蚌埠医学院第一附属医院,安徽蚌埠233004)
唇腭裂是口腔颌面部最常见的先天性畸形,据报道,我国新生儿唇腭裂的发病率约为1.82‰[1]。根据全身是否伴有其他畸形,唇腭裂分为综合征型唇腭裂(SCL/P)和非综合征型唇腭裂(NSCL/P)。SCL/P的病因较为明确,主要是由染色体异常或单基因突变造成的[2,3],而NSCL/P是一种多基因遗传病,由遗传因素和环境因素交互作用引起的[4,5]。NSCL/P具有明显的遗传异质性,但基因的筛选、定位及分析方法仍是其病因学研究的难点和热点。全外显子组测序技术(WES)是利用序列捕获技术将全基因组外显子区域DNA捕捉并富集,然后进行高通量测序的分析方法。WES在筛查范围和检出率等方面较其他测序技术具有明显优势,如对采用Sanger测序和基因芯片测序不能筛查出基因的样本,可采用WES进一步筛查鉴定。国内外学者采用连锁和关联的分析方法陆续报道了多个与NSCL/P有关的候选易感基因和染色体区域[6,7]。2018年1~6月,本研究通过WES精确定位NSCL/P核心家庭的致病基因,旨在为NSCL/P的产前诊断和预防提供科学依据。
1 对象与方法
1.1 研究对象 选择同期蚌埠医学院第一附属医院收治的唇腭裂患者,排除范伍德综合征、歌舞妓面谱综合征、Meckel综合征、腭心面综合征等,诊断为NSCL/P患者6例。所有患者来自不同的核心家庭,父母均为健康人,无先天性疾病。6个核心家庭的基本信息见表1。本研究经蚌埠医学院第一附属医院医学伦理委员会批准,所有研究对象或其监护人知情同意。

表1 6个核心家庭的基本信息
1.2 基因组DNA提取 采集所有家系成员(患者及其父母)肘静脉血,采用基因组DNA提取试剂盒提取基因组DNA,经Qubit3.0荧光定量仪鉴定,所提取的DNA浓度和纯度合格,可用于后续实验。
1.3 全外显子组捕获和测序
1.3.1 全外显子组捕获与建库 按Agilent Sure Select Human All Exon V6试剂盒说明进行全外显子组捕获与建库。将基因组DNA利用Covaris破碎仪随机打断成长度为180~280 bp的片段,经末端修复和加A尾后在片段两端分别连接上接头,制备DNA文库。将pooling后带有特异index的文库与生物素标记的探针液相杂交,再用带链霉素的磁珠捕获基因上的外显子,PCR扩增后进行文库质检,合格即可进行测序。
1.3.2 全外显子组测序 文库构建完成后,采用Qubit3.0荧光定量仪定量DNA浓度合格,采用NGS3K/Caliper鉴定文库的插入片段大小符合预期,采用qPCR法检测文库的有效浓度(3 nmol/L),确保文库质量。文库质量合格后,运用Illumina测序平台测序,测序策略PE150,测序深度100X。
1.3.3 单核苷酸多态性(SNP)/插入缺失(InDel)检测、注释和筛选 获取原始测序数据后,在有参考序列或参考基因组(GRCh37/hg19)的情况下,评估测序质量,包括测序错误率、数据量和比对率等。测序质量合格后检测SNP、InDel、拷贝数变异,并统计和注释检出的变异情况。
1.4 高级分析流程
1.4.1 基于变异有害性的筛选 ①突变位点筛选:去除在千人基因组数据库(1000g_all)、ESP6500数据库(esp6500si_all)、gnomAD数据库(gnomAD_ALL、gnomAD_EAS)中至少有一个频率高于1%的突变,得到可能致病的罕见突变;保留处于编码区或剪接位点区上下10 bp的变异;去除不位于高度保守区且未被软件预测为会影响剪接的同义SNP突变,或处于重复区的小片段(<10 bp)非移码InDel突变;保留被预测为有害或影响剪接的突变。②突变位点有害性分类:根据2015年美国医学遗传学和基因组学学会(ACMG)制定的变异分类系统[8,9],将突变分为致病的、可能致病的、致病性不明确的、可能良性的、良性的,以此来描述孟德尔疾病致病基因中发现的突变。ACMG的变异分类系统中有28个证据类别,根据这28个证据的组合形式进行变异位点的有害性分类。
1.4.2 新生突变位点筛选 新生突变位点筛选方法:①利用基于家系三成员(患者及其父母)的共同分析,采用SAMtools新生突变位点分析方法,得到患者有而其父母没有的新生突变位点,经过突变位点筛选得到最终的候选突变位点;②在得到每个家系成员SNP、InDel等突变位点信息的基础上,筛选患者有而其父母没有的突变位点作为新生突变位点,经过突变位点筛选得到最终的候选突变位点。参照文献[10,11]选择候选突变位点的交集。通过de novo软件检测到的de novo位点数计算新生突变速率。
1.4.3 基于候选基因与疾病表型关系的筛选 采用GO功能富集分析和KEGG通路富集分析确定突变基因参与的最主要生化代谢途径和信号转导途径。
1.4.4 蛋白功能互作网络分析 使用Genemania在线软件GeneMania对候选基因进行蛋白功能互作网络分析。
1.4.5 基因-表型-疾病关联分析 根据提供疾病/表型的名称,通过精准算法,结合测序结果和多种数据库(如DisGeNet),对候选基因筛选排序,构建基因-表型-疾病的关联图。
1.4.6 候选基因疾病相关性排序 综合上述基因-表型-疾病关联分析,针对筛选出的候选基因,依据其与疾病的关联性强弱进行排序。
2 结果
2.1 患者的新生突变情况 见表2。
2.2 GO功能和KEGG通路富集分析结果 GO功能富集分析能够生成细胞组件、生物学途径、分子功能三种结果。其中,富集到生物学途径的突变基因98个,富集到细胞组件的突变基因56个,富集到分子功能的突变基因97个。KEGG通路富集分析显示,从PI3K-AKT信号通路中富集到突变基因6个。
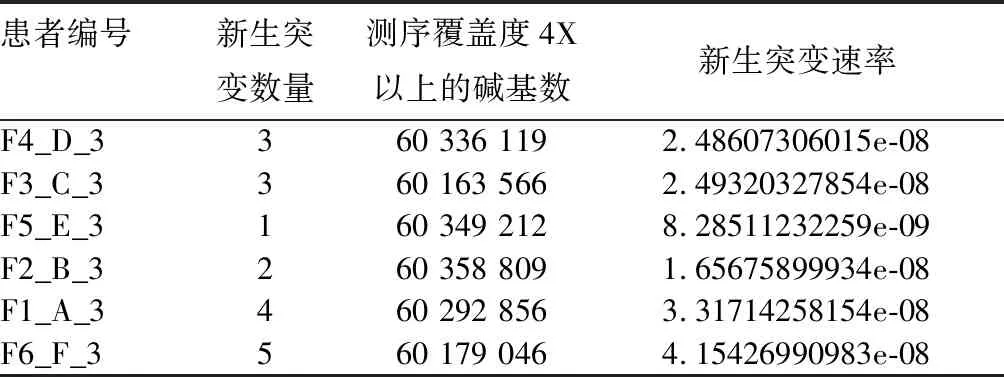
表2 6例患者的新生突变情况
2.3 蛋白功能互作分析结果 见表3。

表3 蛋白功能互作分析结果
注:由于信息量过大,仅取前5行展示。
2.4 候选基因疾病相关性排序 根据筛选出的候选基因,依据其与疾病的关联性强弱进行排序。排序在前五位的基因分别是PLCG2、DNMT3B、L1CAM、TBL1X、NCOR2。因排序只针对筛选出的候选基因,即便排名在第一位,也不能保证关联性一定很强。
2.5 候选基因列表 针对筛选出的候选基因,依据ACMG变异分类标准的SNP新生突变位点可能致病性和SNP或InDel的复合杂合标准再进行筛选,共得到10个候选基因、11个突变位点。见表4。
3 讨论
NSCL/P是一种多基因遗传病,其易感基因较多,不同地区人群的易感基因分布特征亦不完全相同。由于受遗传因素和环境因素交互作用的影响,NSCL/P具有高度遗传变异性、基因微效性等特征,故NSCL/P易感基因的筛选、定位和分析方法要比SCL/P更加复杂[12~15]。WES是利用序列捕获技术将全基因组外显子区域DNA捕捉并富集,然后进行高通量测序的分析方法。在针对孟德尔疾病和复杂疾病的研究中,WES具有明显优势。Basha等[16]通过WES对84例NSCL/P患者的易感基因进行筛查,发现了4个罕见的突变基因,分别是TP63、TBX1、LRP6、GRHL3,表明NSCL/P患者可能仍然携带与SCL/P相关的突变基因。Liu等[17]通过WES对8例NSCL/P患者的易感基因进行筛查,并通过Sanger测序验证,共发现了16个与NSCL/P相关的易感基因。这些研究表明,WES在NSCL/P致病基因的筛查方面具有较高的应用价值。
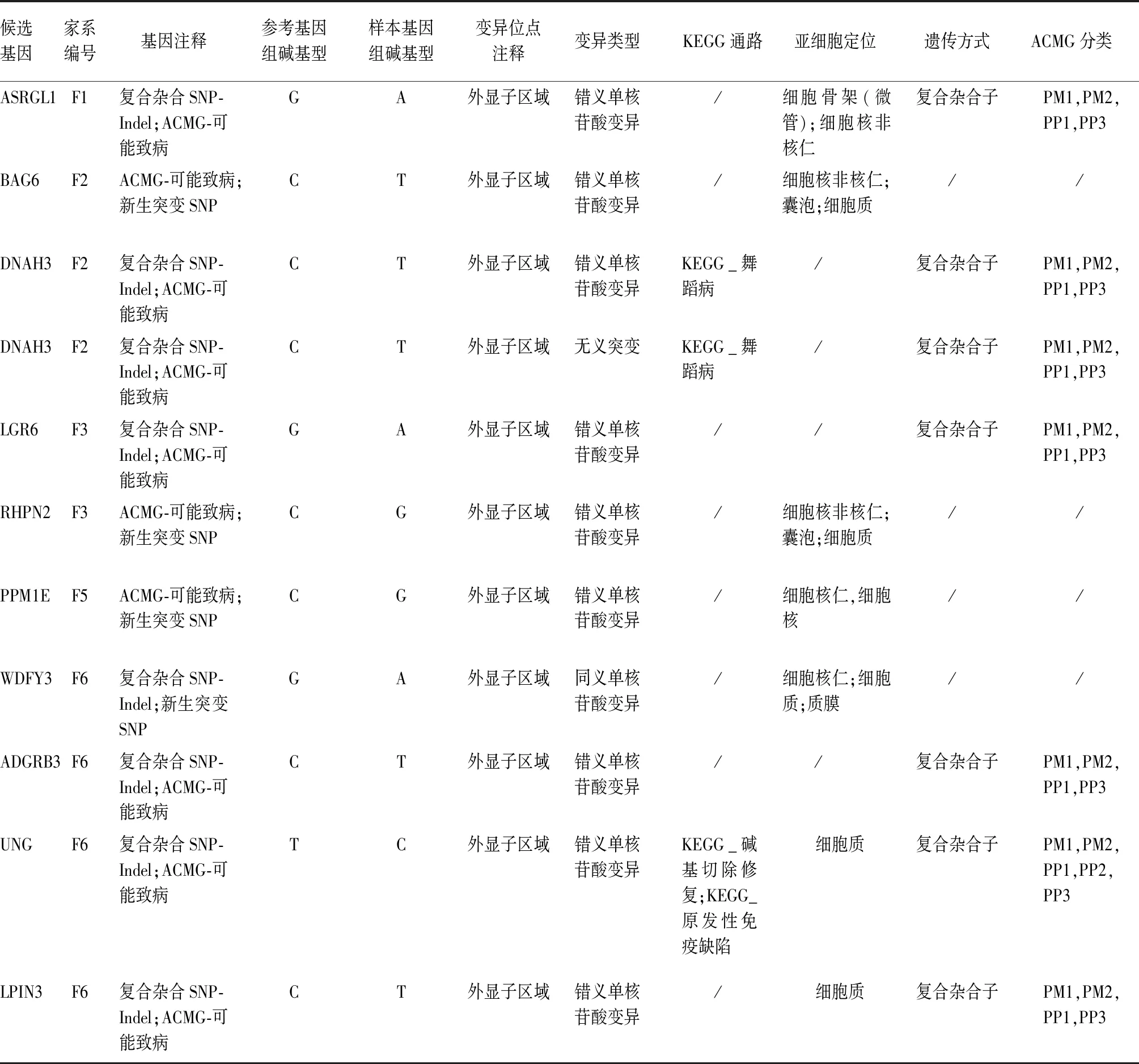
表4 候选基因列表
注:“/”代表未知。
本研究共获得10个候选基因、11个突变位点,这些突变位点均为SNP位点,有无义突变和终止突变,其中WDFY3的SNP位点是同义突变。按ACMG变异分类标准,BAG6、RHPN2、PPM1E和WDRY3四个基因暂无致病等级,剩余基因均有对应的致病等级,其遗传模式为复合杂合。在7个有致病等级的基因中,DNAH3基因的KEGG通路富集分析发现与亨廷顿病有关,UNG基因的KEGG通路富集分析发现与碱基切除修复和初级免疫缺陷有关。但本研究未对筛选到的候选基因和SNP位点进行验证,后续可通过Sanger测序、功能实验等进一步验证。
总之,本研究从6个NSCL/P核心家庭中获得了10个候选基因、11个突变位点,这些候选基因和突变位点可能与NSCL/P发病有关。
