动物病毒宏基因组数据分析平台的建立及应用
2019-11-22史智宾王靖飞
史智宾,王靖飞
(中国农业科学院哈尔滨兽医研究所兽医生物技术国家重点实验室,黑龙江 哈尔滨 150069)
病毒是自然界最丰富的生物体之一,其具有广泛的宿主及复杂的遗传多样性。病毒在微生物生态系统中处于重要的地位,然而在某些环境中病毒微生物群落的全球多样性研究还处于初级阶段[1]。据估计目前对病毒世界的探知还不及1 %[2]。病毒因具有独特的结构及生物学特征,且绝大多数病毒没有专门的宿主细胞系,使得传统的病毒分离鉴定方法只能鉴定出很少一部分病毒。此外,病毒本身也在不断进化以适应宿主环境,增加其核酸多样性,产生新的病毒物种或新的病毒亚型,扩大其宿主范围,从而严重威胁人类的生命健康[3]。传统的病毒分离鉴定技术已无法满足病毒研究的需求,开发研究新的病毒发现技术是研究病毒首要解决的问题。
病毒宏基因组学(Viral metagenomics)技术的出现弥补了病毒研究方面的缺陷。该技术是从环境及其它生物样品中浓缩病毒粒子遗传物质,利用生物信息学分析病毒遗传信息。随着病毒宏基因组学技术的发展,科学家们已经利用该技术获得了成千上万个病毒基因组及大的基因片段[4]。2001年,病毒宏基因组学首次应用于海洋病毒组的研究,在近20年的研究发展中,其应用范围已延伸至土壤、湖水、下水道等无机环境[5-6]以及动物组织、血液、呼吸道、消化道等有机环境[7-9]中。病毒宏基因组学的发展拓展了人类对病毒世界的认知,为新病毒的分离鉴定及未知病毒的发现提供了新的思路[10-12]。近几年病毒宏基因组学技术在兽医研究领域发展迅速,为动物病毒研究提供了便利条件。然而,目前绝大部分病毒宏基因组数据分析经由测序公司完成,商业化分析虽可以保证数据分析的标准化,但其存在成本高昂、耗时长等问题,无法满足及时监控新发疫情时病毒的流行传播情况。因此,低成本、高效率的病毒宏基因组数据分析是目前数据分析工作的重中之重。
本研究建立动物病毒宏基因组数据分析平台,能够高效地进行动物宏病毒组数据分析,能够为动物病毒流行监测及疾病防控提供有效的技术手段。
1 材料与方法
1.1 病毒宏基因组数据分析平台的建立 利用实验室本地linux 服务器建立病毒宏基因组数据分析平台,目前硬件配置:一台安装Red Hat 系统(版本为4.8.5)的Linux 服务器(Linux 版本为3.10.0),该服务器存储容量为1 T,内存48 G,速度266 mHZ。表1所示为数据分析过程中使用软件,所有软件均安装于服务器平台。

表1 主要数据分析软件应用Application数据质量控制Data Quality Control数据预处理Data pre-processing读长注释Read annotation序列拼接及组装效果评价Assembly and assessment重叠序列注释Contig annotation后处理分析Post-processing软件名称Software name FastQC Cutadapt,Bowtie2 BLAST MEGAHIT,QUAST BLAST GenMark,MEGA,BEAST
1.1.1 数据的质量控制及预处理高通量测序后下机得到的数据是长度为150 bp~300 bp 的单端读长数据集或双端读长数据集,称之为原始数据(Raw data)。这样的结果往往会含有质量低的序列或测序过程中插入的接头序列,若不及时处理,会对后续数据分析造成严重的干扰。使用质量控制软件FastQC 对原始数据中接头序列及低质量序列进行检测,然后使用数据处理软件Cutadapt 清洗,同时利用Bowtie2 软件对数据中宿主背景清除,以得到可以用于后续分析的清洁数据集(Clean data)。基于病毒多样性及新病原发现的研究,建立两种数据分析方法,即读长序列分析和重叠序列分析。
1.1.2 读长序列分析读长序列分析方法(Assembly-free methods),是不依赖于序列组装直接利用测序后读长序列进行注释,主要用于样品中病毒微生物的种类组成及多样性研究。其主要利用注释软件BLASTn 对得到的读长序列集进行注释,统计分析读长序列分配至各病毒分类单元情况。
1.1.3 重叠序列分析重叠序列分析方法(Assembly-based methods),是依赖于序列组装,将小而短的读长序列拼接成大而长的重叠序列,利用这些重叠序列进行基因注释,主要用于样品中已知病毒及未知病毒基因序列研究。该方法利用组装软件MEGAHIT 将短的读长序列拼接成长的重叠序列,使用注释软件BLASTn 对得到的重叠序列集进行注释,检测重叠序列与各病毒科病毒序列的同源性。
1.2 临床样品宏基因组分析
1.2.1 样品信息本研究中样品数据为本实验室从安徽省部分猪场采集健康猪鼻拭子后处理的混合样品,经由上海派森诺生物科技有限公司Illumina 测序平台测序所得。
1.2.2 样品数据分析利用质量控制软件检测样品原始数据中存在的接头序列及碱基质量值低的读长,利用数据处理软件对样品数据中接头序列及低质量序列过滤,完成数据过滤后重新进行质量控制,比较过滤前后数据质量,经质量控制检测合格后经下游数据处理分析。读长序列分析直接利用过滤后的读长序列进行注释并统计;重叠序列分析使用序列组装软件对过滤后得到的读长序列进行序列拼接,将拼接后的重叠序列进行基因注释并统计。
2 结 果
2.1 病毒宏基因组数据分析平台的建立 经过长期的数据分析软件筛选和分析方案的优化完善,建立起病毒宏基因组数据分析流程(图1)。该数据分析平台部署了数据质量控制、数据预处理、序列拼接、序列注释等应用程序。根据动物新病毒发现中对敏感性和特异性的不同需求,建立了两种基因序列注释策略:读长注释分析和重叠序列分析。前者能提高注释的敏感性,有利于发现低拷贝的病毒基因组序列;后者可提高注释的特异性,保障注释结果的准确性。可以根据不同研究需要选择不同的分析方法。
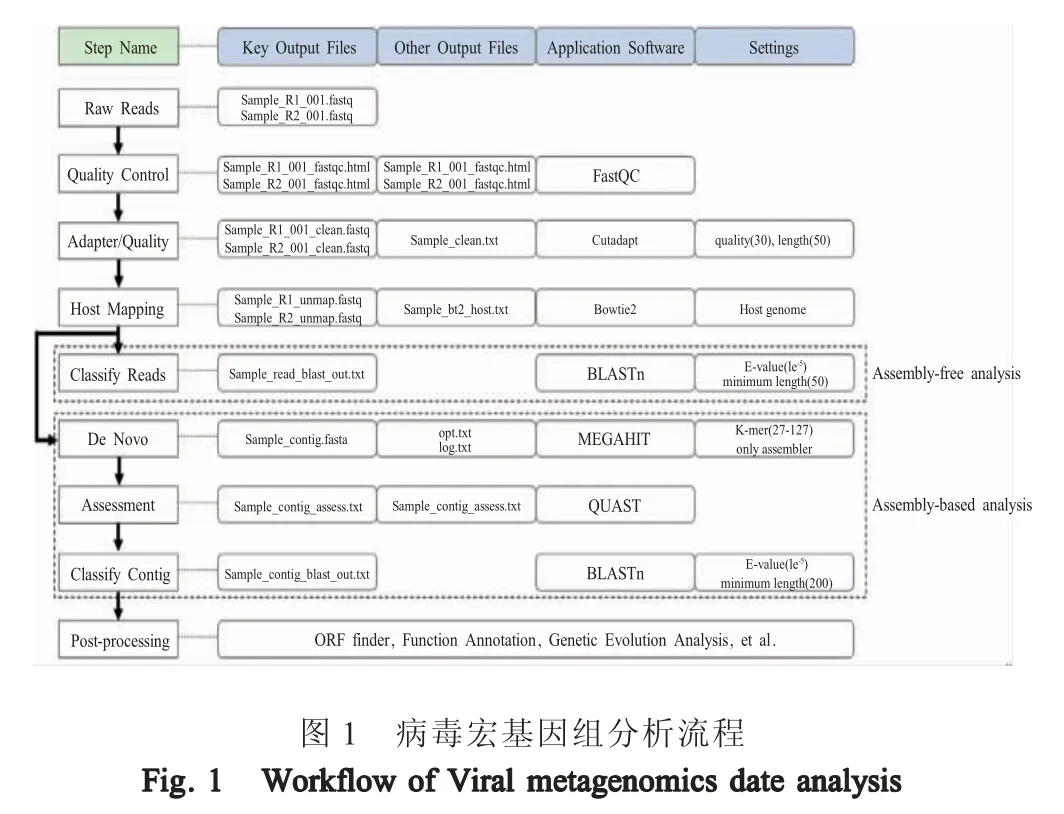
2.2 临床样品的宏基因组分析
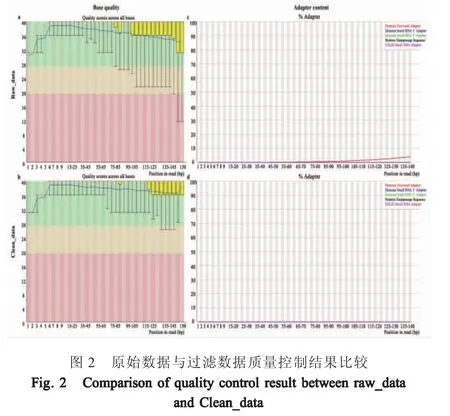
2.2.1 样品数据过滤及质量控制利用数据质量控制软件FastQC 对原始测序数据进行质量控制检测。碱基质量分布图中蓝色线表示各位置碱基质量平均数,一般平均数高于25,所有位置的10 %中位数大于20 表示测序质量结果较好。接头序列分布图中主要对4 种通用测序接头序列进行检测,根据测序数据中接头序列的存在情况进行处理。原始读长序列的3' 末端碱基质量值偏低(图2a)并包含有Illumina 测序通用接头序列(图2c)。利用数据处理软件Cutadapt[13]进行数据接头序列及低质量序列的去除,过滤后重新进行质量控制检测。处理后所有读长序列各位置碱基质量均高于35,且所有位置10 %中位数大于26 (图2b),并且数据集中所含有的接头序列已被去除干净(图2d)。上述结果表明经处理后得到的数据可以进行下游分析。
2.2.2 序列组装效果评价利用本地化安装的组装效果评价软件对得到的重叠序列集进行评价,A 为本地化建立的病毒宏基因组数据分析方法组装后得到重叠序列集的组装效果评价结果,B 为经公司测序分析后得到的重叠序列集的组装效果评价结果(表2)。N50 作为组装效果评价中的重要指标,其是将拼接得到的重叠序列由长到短进行排序并累加,当累加和达到重叠序列总长度的50 %时,最后参与加和的那一条重叠序列即为拼接后重叠序列数据集的N50 的长度。结果A 与结果B 的N50 长度分别为1 052 bp 和1 002 bp,表明二者之间在序列拼接方面无明显差异。
2.2.3 基因注释测序后共计产生32233976 条读长,每条读长序列长度为150 bp,经过接头序列及低质量序列去除后得到31490033 条读长。读长序列分析注释后发现共有742196 条读长与病毒序列有较高的相似性,占总体读长序列数据的2.36 %。重叠序列分析得到415 117 条重叠序列,其中307 条被注释到病毒基因组包括指环病毒科(Anelloviridae)、沙粒病毒科(Arenaviridae)、星状病毒科(Astroviridae)、环状病毒科(Circoviridae)、冠状病毒科(Coronaviridae)、细小病毒科(Parvoviridae)、小 RNA 病毒科(Picornaviridae)、痘病毒科(Poxviridae)、Smacoviridae、Genomoviridae、逆转录病毒科(Retroviridae)和副粘病毒科(Paramyxoviridae)等12 个病毒科的64 种病毒(表3)。从注释结果看,该宏病毒组数据注释到冠状病毒科的多个种属(同源性为70 %~100 %),其中与猪德尔塔冠状病毒(Porcine deltacoronavirus)的同源性最高(99.775 %)。读长序列分析耗时1.5 d,重叠序列分析耗时3.5 d,表明本地化建立的动物病毒宏基因组数据分析平台能够高效的完成宏病毒组数据分析工作。
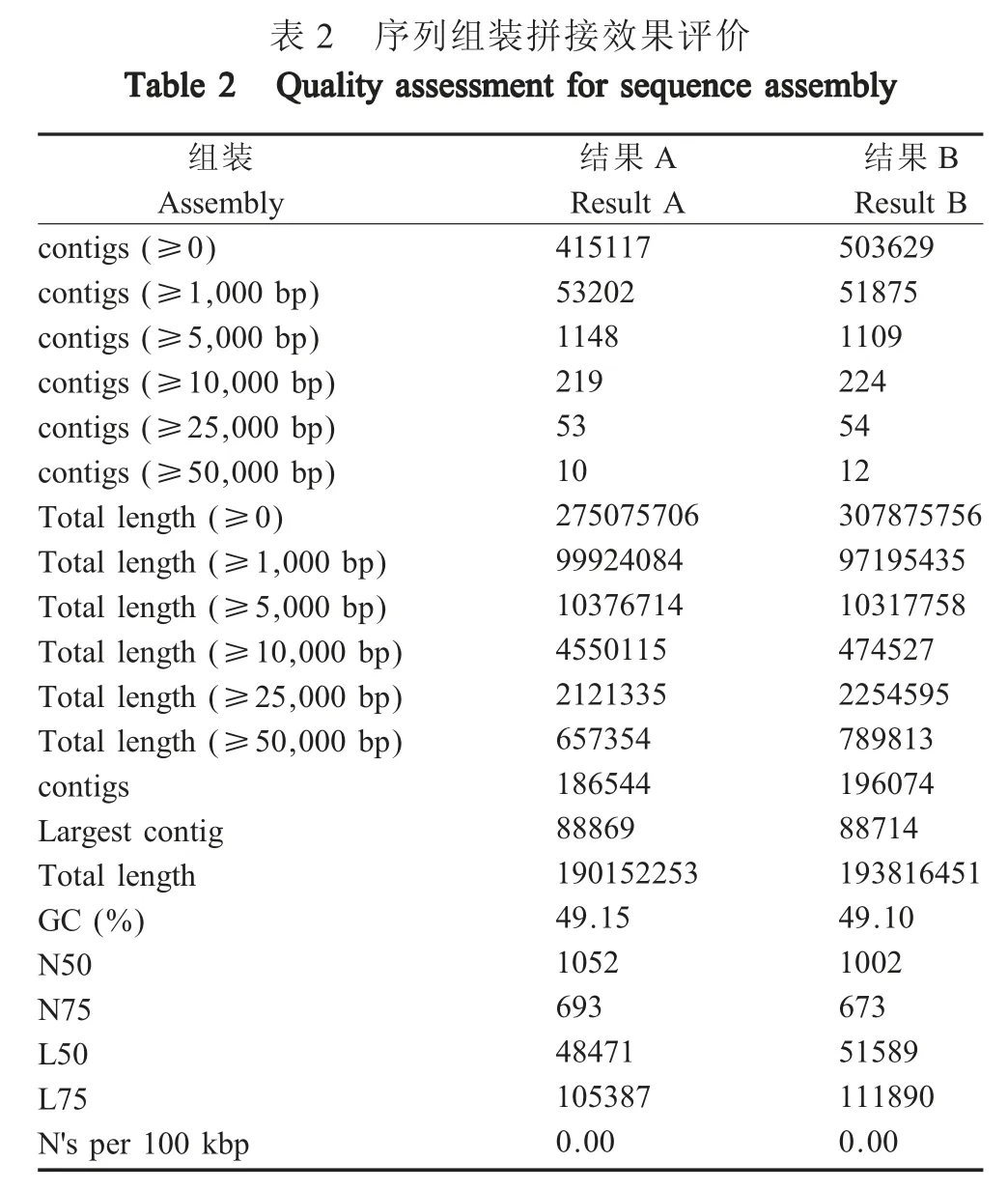
表2 序列组装拼接效果评价组装Assembly contigs (≥0)contigs (≥1,000 bp)contigs (≥5,000 bp)contigs (≥10,000 bp)contigs (≥25,000 bp)contigs (≥50,000 bp)Total length (≥0)Total length (≥1,000 bp)Total length (≥5,000 bp)Total length (≥10,000 bp)Total length (≥25,000 bp)Total length (≥50,000 bp)contigs Largest contig Total length GC (%)N50 N75 L50 L75 N's per 100 kbp结果A Result A 415117 53202 1148 219 53 10 275075706 99924084 10376714 4550115 2121335 657354 186544 88869 190152253 49.15 1052 693 48471 105387 0.00结果B Result B 503629 51875 1109 224 54 12 307875756 97195435 10317758 474527 2254595 789813 196074 88714 193816451 49.10 1002 673 51589 111890 0.00
3 讨 论
本研究利用本地化建立的动物病毒宏基因组学数据分析平台对猪源鼻拭子宏病毒组数据进行了分析,结果表明该样品中包含大量指环病毒、环状病毒、冠状病毒和细小病毒等。有研究发现其它动物如熊猫[14]、蝙蝠[15]等体内也同样检测到有上述病毒的存在,表明这些病毒可能在动物体内普遍存在。同时,在该样品中还发现有微量猪星状病毒、捷申病毒和白血病病毒等,这也揭示了猪体内丰富的病毒多样性。因此,本地化建立的数据分析平台能够满足动物病毒宏基因组数据的分析。
本研究建立的动物病毒宏基因组数据分析平台融合了数据质控、数据过滤、序列拼接和基因注释等多种分析手段。与之前文献及测序公司已有的分析平台相比,该分析平台所选择分析软件兼容性强,分析效率更高,分析流程相对简单,在本地化平台能够高效的完成数据分析工作。同时,本研究对两种序列分析方法进行比较,发现二者在数据分析中各存在优劣势。在微生物群落综合性与复杂性分析方面,读长序列分析拥有着更大的优势,其能够对病毒微生物群落功能与结构进行聚类分析,若能有足够的测序深度和参考数据库的覆盖范围,该方法可以对任意复杂的病毒群落进行分析;而重叠序列分析只有在基因组有足够的丰度时才能够完成多个基因组的构建,且在复杂的病毒微生物群落中只有少部分基因组可以通过组装得到分析。此外,在计算成本方面,重叠序列分析需要高昂的计算成本来进行基因组的组装及注释。但相比较而言,重叠序列分析在新病毒的发现及整合微生物基因组学方面更具优势。重叠序列分析能够对参考基因库无亲缘关系的基因组序列进行解析,而读长序列分析无法分辨未知基因组序列;重叠序列分析可以利用得到的基因组片段信息支持纯培养分离基因组分析,而读长序列分析无法完成[16];重叠序列分析在新病毒的发现[17]及病毒的遗传进化分析方面应用较广。此外,将原始读长序列转化为有意义的微生物特征分析工具也在持续不断的更新改进,基于读长序列的病毒株水平分析目前也已被应用[18-20]。
值得注意的是,不论哪种序列分析方法,都依赖于病毒微生物群落的组成及复杂性、测序的深度、测序数据集的大小和计算分析资源的可用性。因此,在实际的样品数据分析时需要考虑各种因素对数据分析及结果的影响,同时利用两种序列分析方法可以做到相互验证及补充,更能保证结果的可靠性及准确性。
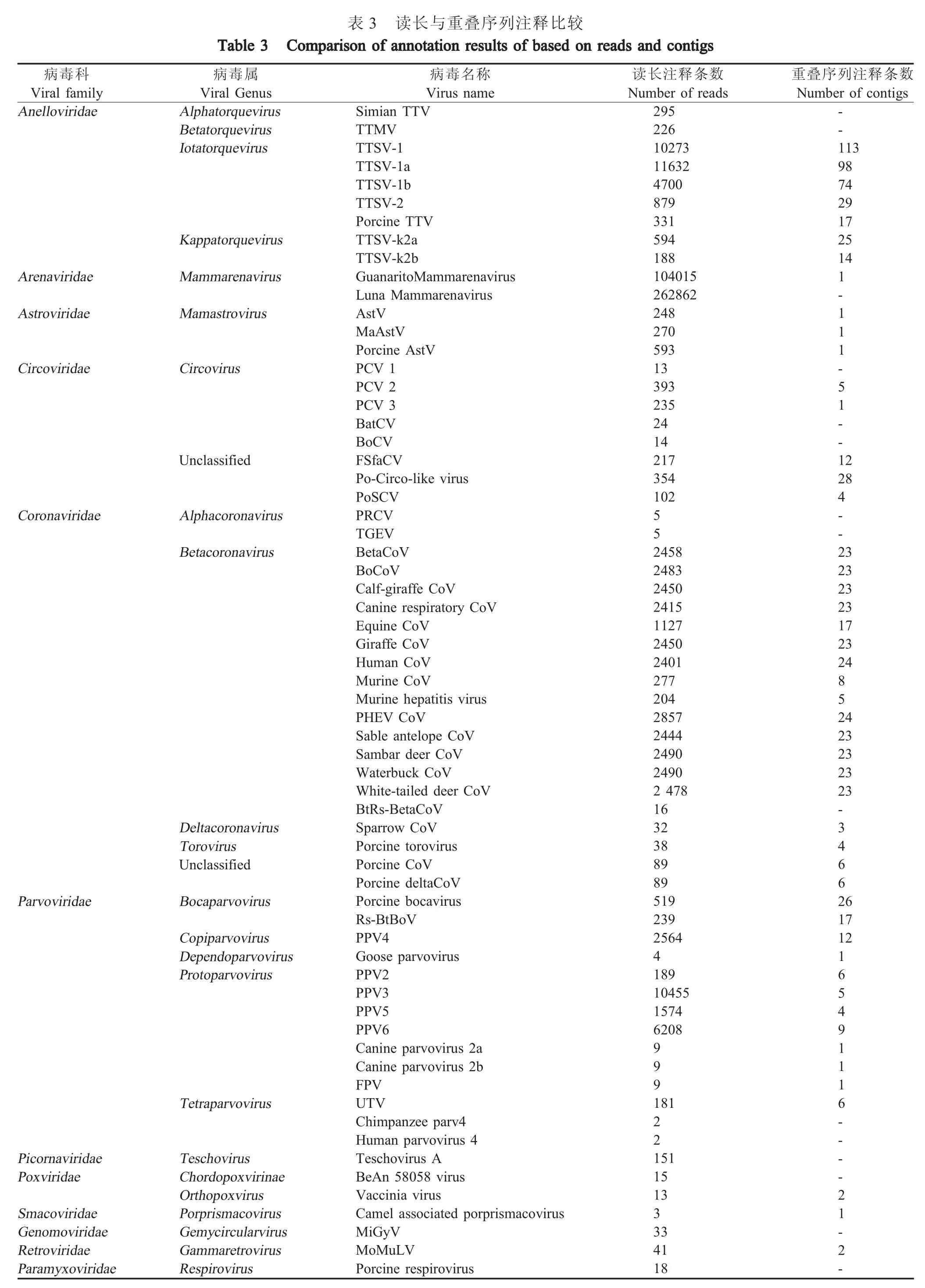
病毒科Viral family Anelloviridae Viral Genus Alphatorquevirus Betatorquevirus Iotatorquevirus序列注释条数Number of contigs--113 98 74 29 17 25 14 Arenaviridae Astroviridae Kappatorquevirus Mammarenavirus Mamastrovirus Circoviridae Circovirus Unclassified Number of reads 295 226 10273 11632 4700 879 331 594 188 104015 262862 248 270 593 13 393 235 24 14 217 354 102 1-111-51--1 2 28 Coronaviridae Alphacoronavirus Betacoronavirus 55 4--2 3 23 23 23 17 23 24 852 4 23 23 23 23 Parvoviridae Deltacoronavirus Torovirus Unclassified Bocaparvovirus Copiparvovirus Dependoparvovirus Protoparvovirus 2458 2483 2450 2415 1127 2450 2401 277 204 2857 2444 2490 2490 2 478 16 32 38 89 89 519 239 2564 4 189 10455 1574 6208-34662 6 Virus name Simian TTV TTMV TTSV-1 TTSV-1a TTSV-1b TTSV-2 Porcine TTV TTSV-k2a TTSV-k2b GuanaritoMammarenavirus Luna Mammarenavirus AstV MaAstV Porcine AstV PCV 1 PCV 2 PCV 3 BatCV BoCV FSfaCV Po-Circo-like virus PoSCV PRCV TGEV BetaCoV BoCoV Calf-giraffe CoV Canine respiratory CoV Equine CoV Giraffe CoV Human CoV Murine CoV Murine hepatitis virus PHEV CoV Sable antelope CoV Sambar deer CoV Waterbuck CoV White-tailed deer CoV BtRs-BetaCoV Sparrow CoV Porcine torovirus Porcine CoV Porcine deltaCoV Porcine bocavirus Rs-BtBoV PPV4 Goose parvovirus PPV2 PPV3 PPV5 PPV6 Canine parvovirus 2a Canine parvovirus 2b FPV UTV Chimpanzee parv4 Human parvovirus 4 Teschovirus A BeAn 58058 virus Vaccinia virus Camel associated porprismacovirus MiGyV MoMuLV Porcine respirovirus 17 12 999 Tetraparvovirus 181 22 Picornaviridae Poxviridae Smacoviridae Genomoviridae Retroviridae Paramyxoviridae Teschovirus Chordopoxvirinae Orthopoxvirus Porprismacovirus Gemycircularvirus Gammaretrovirus Respirovirus 151 15 13 3 33 41 18 165491116----21-2-
本地化建立的动物病毒宏基因组数据分析平台,能够高效的进行动物宏病毒组数据分析,这对于新疫情暴发时及时确定病因,提出解决措施意义重大。然而,目前本地化建立的病毒宏基因组学数据分析平台还处于初步应用阶段,未来数据分析过程中在保证工作与时间效率的同时,还需优化结果的报告形式及可视化展示,实现高标准化的数据分析,更好的应用于动物病毒宏基因组数据分析。该病毒宏基因组数据分析平台的建立为潜在致病原及新病原的发现提供便利工具,为全国流行病学分析及疾病防控研究提供技术支持。
