精神分裂症患者肠道菌群结构特征初步观察与分析☆
2019-08-22黄侠卓敏李时佳吴逢春陈军韩俊南吴凯
黄侠 卓敏 李时佳 吴逢春 陈军 韩俊南 吴凯*※
近年来,由于肠道菌群与人类健康和疾病的关系密切而备受关注。肠道菌群不仅可调控食物消化能力,维持免疫系统的运作,而且能通过脑—肠—微生物轴影响大脑的活动[1-3]。已有动物研究表明,改变肠道菌群组成能诱导动物产生类似精神类疾病的症状和行为[4-5],并且在动物模型中已证实肠道菌群改变与孤独症存在关联[6-7]。但是,有关人类精神分裂症与肠道菌群相关性的研究仍少有报道[8-10],且肠道菌群是个复杂且庞大的生态系统,不同地区、不同年龄阶段人群其肠道菌群都存在一定差异。因此,本研究对精神分裂症患者肠道菌群进行16S rRNA基因高通量测序,探索其肠道菌群特征,及可能的病理机制,为精神分裂症的临床诊断和治疗提供参考依据。
1 对象与方法
1.1 研究对象选取2017年4月至2017年12月就诊于广州市惠爱医院门诊及住院的精神分裂症患者作为患者组。入组标准:①符合《美国精神障碍诊断与统计手册第四版》(Diagnostic and Statistical Manual of Mental Disorders,Fourth Edition,DSM-Ⅳ)精神分裂症的诊断标准;②年龄18~45岁;③病程≥2年,目前使用的抗精神病药物种类和剂量在8周内稳定;④血常规检查无异常;⑤无精神活性物质滥用或依赖(酒精、咖啡因、尼古丁除外)。排除标准:①患有糖尿病、甲状腺疾病、高血压、心脏病等可能影响肠道菌群稳定性的疾病;②近3个月内有腹泻;③近3个月内使用过抗生素类或益生菌类药物;④妊娠、哺乳期或月经期妇女;⑤合并其他符合DSM-Ⅳ诊断标准的精神疾病。共收集16例患者。
对照组来自同一时间段内在广州大学城公开招募的健康志愿者。入组标准:①不符合DSM-Ⅳ任何精神疾病的诊断;②无精神疾病史及家族史;③年龄18~45岁;④血常规检查无异常;⑤无精神活性物质滥用或依赖 (酒精、咖啡因、尼古丁除外)。排除标准同患者组。共收集20名对照者。
本次研究经过广州市惠爱医院伦理委员会批准。所有受试者签署知情同意书。
1.2 研究方法
1.2.1 DNA提取和16S rRNA测序 采集所有受试者的新鲜粪便样本于50 mL无菌采便盒内,-80℃保存。根据TIANamp Stool DNA Kit(天根生化科技有限公司)说明书提取总DNA,用NanoDrop 2000c(Thermo Scientific)测定DNA的浓度和纯度,1%琼脂糖凝胶电泳检测DNA的完整性。以细菌16S rRNA基因的V3可变区为目标片段,扩增DNA构建测序文库,使用Ion PGMTM测序仪(博奥木华基因科技有限公司)进行测序。
1.2.2 生物信息学分析 采用 Qiime(Quantitative Insights into Microbial Ecology)1.8.0 软件 (http://qiime.org/)对原始序列进行优化处理,得到高质量序列[11]。以UCLUST算法为基础,将高质量序列以97%核苷酸序列相似性聚类成可操作分类单元(operational taxonomic units,OTUs), 应用 Green-Gene数据库(version 13.8)对OTUs进行物种信息注释。基于OTUs的注释信息,计算α多样性(Chao1指数、Ace指数、Shannon指数和 Simpson指数)。根据细菌的门和属两个水平上序列数的统计信息,将各物种丰度归一化到同一数量级,用log2(患者组/对照组)计算样品之间物种丰度的倍数差异,进而分析不同菌群相对丰度的组间差异[12]。采用线性判别效应分析(linear discriminant analysis effect size,LEfSe)描述两组间具有统计学差异的菌群标志物,并用线性判别分析(linear discriminant analysis,LDA)估计每个差异菌群的效应大小,本研究LDA判别界值取1.5。为深入了解肠道菌群在两组中发挥的生物学作用,利用PICRUSt软件,结合GreenGene数据库和KEGG数据库,对16S rRNA数据进行功能谱预测。
1.3 统计学分析统计学分析使用SPSS 23.0。两组被试年龄、肠道菌群α多样性指数等正态分布的数据采用均数±标准差(±s)描述,组间比较采用独立样本t检验。性别等组间比较采用Fisher精确检验。门、属水平上的肠道菌群相对丰度呈偏态分布,采用中位数(下四分位数,上四分位数)[M(QL,QU)]描述,过滤掉在少于 20%被试中存在的OTUs后,采用Wilcoxon秩和检验进行组间比较。检验水准α=0.05,双侧检验。
2 结果
2.1 人口学资料患者组年龄(33.63±8.04)岁,男性 7例,女性 9例;对照组年龄(27.60±7.07)岁,男性16名,女性 4名。两组年龄(t=-2.391,P=0.022)、性别(P=0.038)差异有统计学意义。患者组的病程为(5.19±3.76)年,发病年龄为(28.44±7.44)岁,使用抗精神病药物为奥氮平、利培酮、舒必利中两种或三种。
2.2 菌群多样性分析对36例粪便样本DNA进行测序,共获得12436779条高质量序列,其中患者组6556239条。通过序列相似性聚类,得到5475个 OTUs,其中患者组4513个,对照组 4545个,3583个为两组共有。α多样性分析表明,两组的 Chao1 指数(t=-1.056,P=0.298)、Ace 指数(t=-1.254,P=0.218)、Shannon 指数(t=0.070,P=0.945)和 Simpson 指数(t=0.155,P=0.878)均没有统计学差异(表 1)。
2.3 菌群结构分析在门水平上,对照组的优势菌群主要属于厚壁菌门 (Firmicutes)和拟杆菌门(Bacteroidetes),占总细菌的93.73%,梭杆菌门(Fusobacteria)、变形菌门(Proteobacteria)和放线菌门(Actinobacteria)合占1.99%,其它细菌占剩下的4.28%。与对照组相比,患者组的优势菌群未改变,厚壁菌门和拟杆菌门占总细菌的90.60%,厚壁菌门与拟杆菌门的相对丰度比值、梭杆菌门与变形菌门相对丰度比值较对照组略变大,放线菌门的相对丰度有所增加,但变化均无统计学意义(P>0.05)。低丰度的TM7门菌群在患者组中的相对丰度较对照组增加,且差异有统计学意义(Z=-2.042,P=0.041)(表 2)。

表1 精神分裂症患者与对照肠道菌群α多样性分析

表2 精神分裂症患者与对照门水平上的肠道菌群相对丰度及其比值
在属水平上,对照组粪便中占主导地位菌属有拟杆菌属(Bacteroides,21.02%)、普雷沃氏菌属(Prevotella,10.43%)、粪杆菌属(Faecalibacterium,6.58%)和巨单胞菌属(Megamonas,1.36%),而患者组以拟杆菌属(34.39%)、粪杆菌属(4.35%)、梭杆菌属 (Fusobacterium,1.09%)、 巨单胞菌属(0.23%)为优势菌属(图1,表3)。与对照组相比,患者组有8个菌属的相对丰度均升高,差异具有统计学意义(P<0.05)。其中5个属于放线菌门,分别为放线菌属(Actinomyces)(Z=-2.010,P=0.044)、罗氏菌属 (Rothia)(Z=-2.702,P=0.007)、 斯卡多维亚氏菌属(Scardovia)(Z=-2.701,P=0.007)、奇异菌属(Atopobium)(Z=-2.076,P=0.038)和伊格尔兹氏菌属 (Eggerthella)(Z=-2.019,P=0.043);3 个属于厚壁菌门,分别为乳杆菌属(Lactobacillus)(Z=-2.570,P=0.010)、Moryella(Z=-3.014,P=0.003)和G07菌属(Z=-1.983,P=0.047)(表 3)。进一步LEfSe分析发现,以上8个组间差异菌属中,除罗氏菌属和伊格尔兹氏菌属之外,其余6个在两组间具有统计学差异(P<0.05)(图 2A)。
此外,在科水平上,患者组中消化链球菌科(Peptostreptococcaceae)、乳杆菌科(Lactobacillaceae)、放线菌科(Actinomycetaceae)3个科的相对菌群丰度高于对照组 (P<0.05),而组织菌科(Tissierellaceae)的相对丰度低于对照组(P<0.05)(图 2B)。
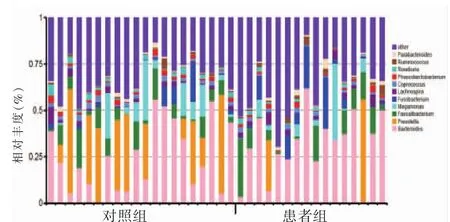
图1 对照组和患者组在属水平上肠道主要菌群组成分布图
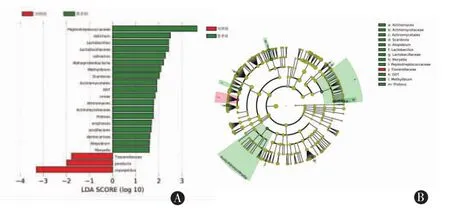
图2 对照组和患者组肠道菌群组成的LEfSe分析。A为仅对LDA≥1.5的分类群进行可视化;B为两组间差异最大的菌群
2.4 菌群功能分析功能分析结果显示患者组和对照组有多种生物学途径存在差异 (P<0.05),包括β-丙氨酸、核黄素、磷酸盐、脂肪酸和丙酮酸等代谢途径,苯甲酸、二甲苯、二噁英、萘、氯烷烃和氯烯烃等降解途径,四环素和脂肪酸生物合成途径,糖酵解/糖异生、戊糖磷酸途径等(图3)。
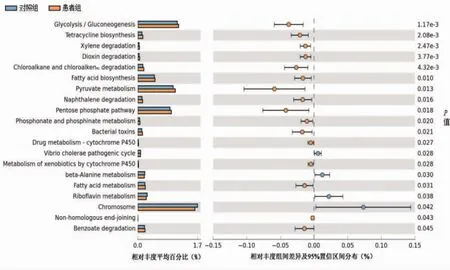
图3 对照组和患者组肠道菌群功能预测分析。图中为相对丰度在两组之间差异有统计学意义的生物学途径(P<0.05),右侧P值为校正后的值
3 讨论
健康人肠道菌群主要由厚壁菌门(60%~80%)、拟杆菌门(20%~30%)、放线菌门(<10%)和变形菌门(<1%)组成[13-18]。本研究也发现,对照组中相对丰度较高的门类为厚壁菌门(46%)和拟杆菌门(47%),只是两者的占比稍有变化,可能是本研究样本量较小,不足以显示其分布状况,也可能与受试者的饮食习惯和地理环境等因素有关。本研究中,与对照组相比,患者的肠道菌群发生改变:在门水平,TM7门菌群相对丰度偏高;在科水平,消化链球菌科、乳杆菌科和放线菌科的菌群相对丰度偏高,而组织菌科的相对丰度偏低;在属水平,6个菌属的相对丰度均偏高,其中3个属于放线菌门(放线菌属、奇异菌属和斯卡多维亚氏菌属),3个属于厚壁菌门 (乳杆菌属、Moryella和G07菌属)。SCHWARZ等[19]研究发现首发精神病患者粪便中乳杆菌属的相对丰度比健康对照组高,且乳杆菌属丰度与症状的严重程度呈正相关;此外,SHEN等[9]发现在精神分裂症缓解期患者肠道中乳杆菌属的相对丰度也高于健康对照组。本研究与这些文献结果一致,同时也提示乳杆菌属相对丰度升高与精神分裂症的发生发展有一定相关性[20]。总的来说,本研究发现精神分裂症患者肠道菌群较健康对照者发生改变,这些变化的菌群可能在精神分裂症发病中发挥重要生物学作用,有望为精神分裂症的临床诊断和治疗提供一定参考。
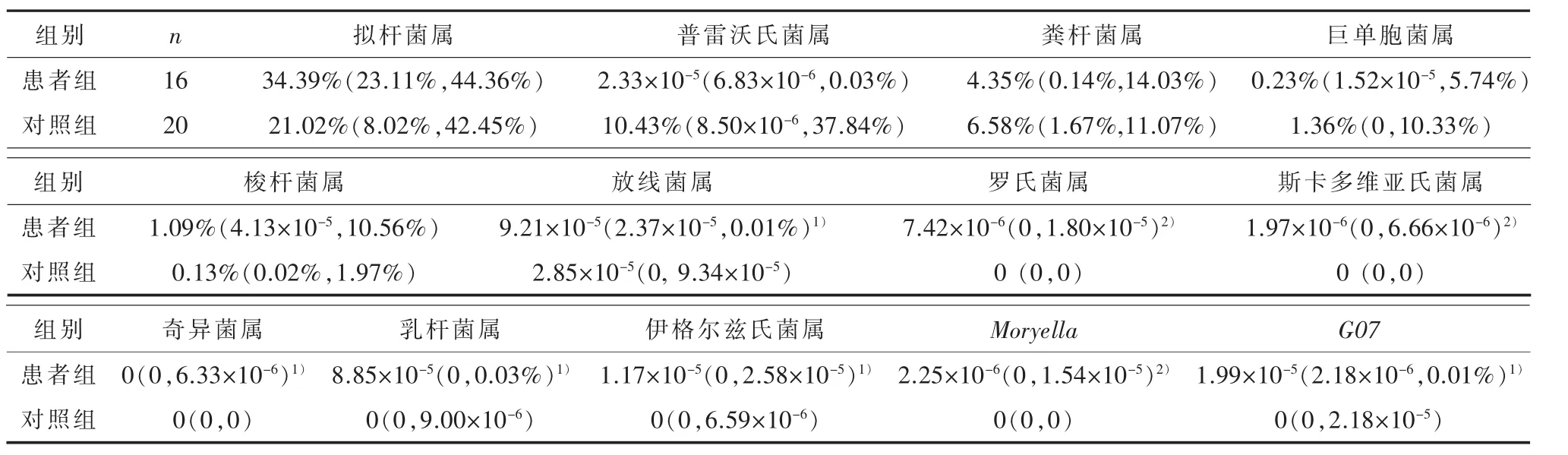
表3 精神分裂症患者与对照属水平上的肠道菌群相对丰度
大脑的认知功能与其能量供应能力协同进化,精神分裂症患者存在认知功能障碍,其大脑中能量代谢存在异常[21-22]。对精神分裂症患者尸检的研究将大脑前额叶皮质组织样本进行转录、蛋白质和代谢组学分析发现,脂肪酸β-氧化酶的转录水平显著增加[23],而血清和尿液分析推测脂肪酸代谢途径在患者中占主要地位[24]。本研究发现精神分裂症患者体内脂肪酸代谢和丙酮酸代谢途径与健康对照存在差异。丙酮酸合成、乙酰CoA合成和脂肪酸生物合成是合成短链脂肪酸(short chain fat acid,SCFAs)的三种基本反应。HE 等[10]通过分析受试者肠道微生物群的组成和脑内细胞膜破坏的标志物(即胆碱),得到精神分裂症高风险和超高风险受试者脑—肠—微生物轴的特征,其中,超高风险受试者肠道中乳杆菌属相对丰度增加可以刺激肿瘤坏死因子 (tumor necrosis factor,TNF)的产生,本研究中也发现患者组乳杆菌属水平增加。另外,SCFAs可与体内TNF穿过血脑屏障进入中枢神经系统,激活小胶质细胞,干扰神经细胞的膜代谢,进而诱发精神分裂症[7,25]。据此,推测肠道微生物变化可能引起机体能量代谢异常,菌群代谢物异常,诱导炎症因子水平改变,进而引起精神分裂症发生。基于此,未来可以扩大样本量进行纵向深入的研究,建立肠道微生物与血清代谢物相结合的预测模型,用于精神分裂症的临床辅助诊断和治疗。
本研究分析精神分裂症患者和健康对照者肠道菌群的差异,为精神分裂症的临床诊断和治疗提供一定参考。但也存在以下局限:样本量较小,未考虑饮食习惯、生活方式等对肠道菌群的影响等,此外,本研究中的患者在入组时已经服用抗精神病药物,无法分析患者用药前后肠道菌群变化情况,不能明确抗精神病药物对肠道菌群有何影响。后续需要扩大样本量,纳入患者详细的临床信息,尽可能控制干扰因素进行纵向深入研究;针对首发未用药患者进行长期随访,研究用药前后患者肠道菌群变化情况;对关键菌株进行分离和鉴定,并通过疾病动物模型等研究其在精神分裂症中的潜在作用机制。
