可待因的人体代谢及药物相互作用研究综述
2019-06-13李倩倩林忠泽张清民吴盼盼李冬利
李倩倩,林忠泽,张清民,吴盼盼,李冬利
(1.特一药业集团股份有限公司,广东 江门 529200; 2.五邑大学 生物科技与大健康学院, 广东 江门 529020)
1 理化性质
可待因的分子式为C18H21NO3,在鸦片中的含量占0.7%~2.5%。磷酸可待因的分子式为C18H24NO7P,为白色针状结晶或晶体粉末,无臭,光照可变性,易溶于水,微溶于乙醇,其水溶液呈酸性[1]。
2 可待因的药物代谢动力学研究
2.1 口服给药代谢过程
可待因被广泛用于止咳药和镇痛药,在人体肝脏中通过基因多肽酶细胞色素P450 3A4(CYP3A4)的作用发生N-脱甲基生成去甲可待因、进行葡萄糖醛酸化生成可待因-葡萄糖醛酸结合物(C6G),这是可待因体内代谢的主要途径(超过80%)[2],但是代谢产物C6G、去甲可待因及去甲可待因-葡萄糖醛酸结合物(NCG)都不具备药理活性。可待因在体内代谢的次要途径是通过基因多肽酶细胞色素P450 2D6(CYP2D6)的作用发生O-脱甲基而代谢为吗啡(低于10%),吗啡进一步代谢为吗啡-3-葡萄糖醛酸结合物(M3G)和吗啡-6-葡萄糖醛酸结合物(M6G),吗啡和M6G具有阿片活性,是可待因产生镇痛、止咳和止泻作用的基本方式[3]。目前可待因有7种已知代谢产物(如图1所示):C6G、吗啡、M3G、M6G、NCG、去甲吗啡和去甲可待因[4]。
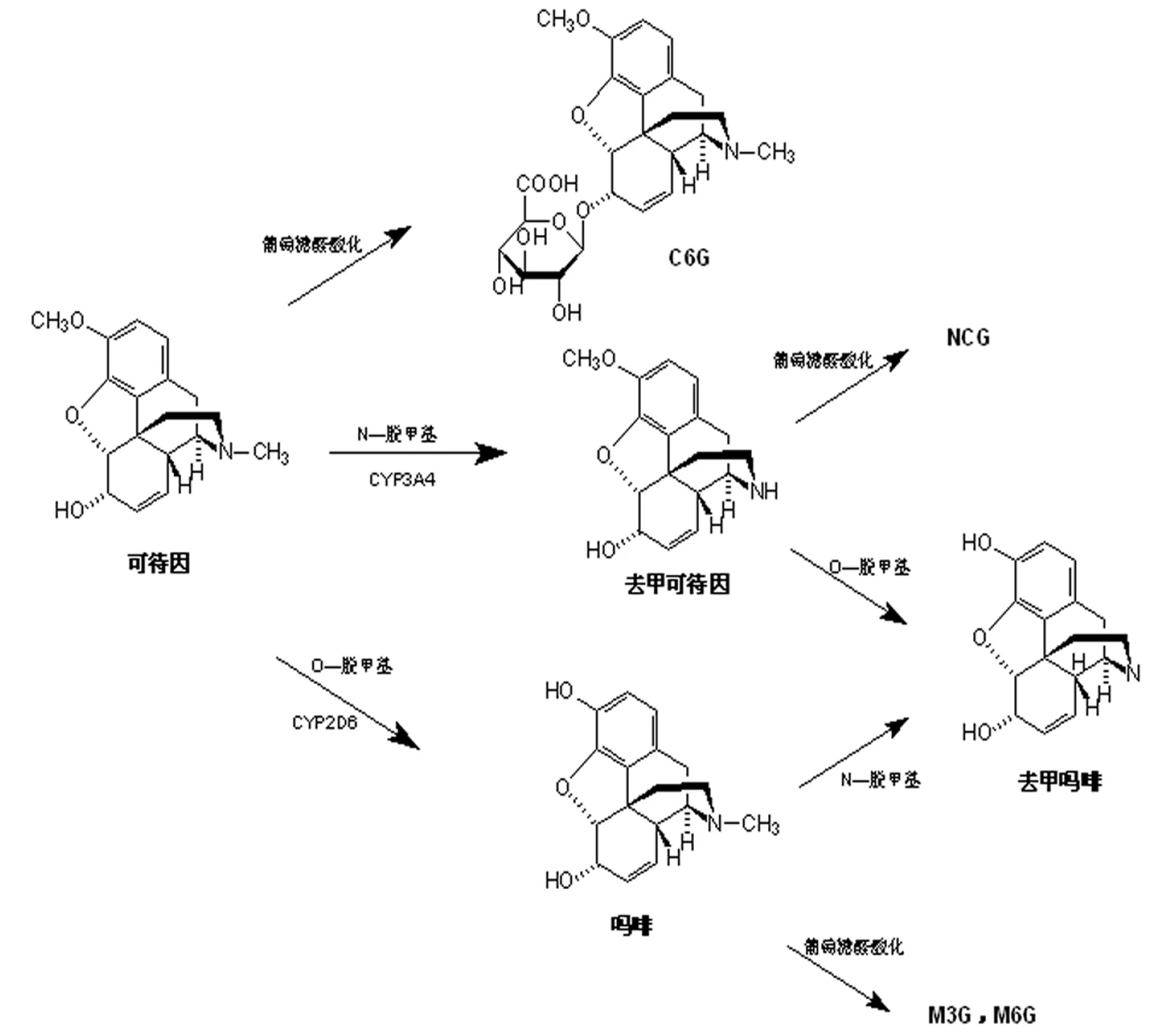
图1 可待因的7种代谢途径
2.1.1 CYP2D6代谢酶的统计学分析
目前从未发现CYP3A4对任何药物的代谢存在个体差异,但是不同亚型的CYP2D6对药物代谢的转化率具有明显差异[5]。CYP2D6具有基因多态性,存在超过80余种不同的等位基因,并且在不同族群中的分布差异很大[6]。按照CYP2D6的活性大小,人类可分为超速代谢型(UMs)、正常代谢型(NMs)、中间代谢型(IMS)和弱代谢型(PMs)[7]。
根据Andrea Gaedigk等人的统计分析[7],全球范围内超速代谢型的比例为1%~21%,正常代谢型的比例为67%~90%,中间代谢型的比例为0.4%~11%,弱代谢型人的比例为0.4%~5.4%。但是不同族群中比例不同,其中东亚人群中超速代谢型的比例为1.4%,弱代谢型的比例为0.4%。
2.1.2 可待因及其葡萄糖醛酸代谢物
根据Z.R.CHEN[4]对磷酸可待因片剂的人体代谢动力学研究,单次口服磷酸可待因片剂后,可待因和C6G的达峰时间、半衰期接近,分别为1h和3.2h,而C6G的AUC值比可待因高15.8倍。可待因的平均肾脏清除速率是183mL/min,与尿液pH值存在反比关系;C6G的平均肾脏清除速率是55mL/min,与尿液pH值无关。给药后48h内大约86%的药物可通过尿液排泄,其中60%为C6G形式,7%为吗啡形式(包括M3G和M6G),7%为去甲可待因形式,12%为可待因原形。
重复多次给药后,可待因和C6G的达峰时间、半衰期、肾脏清除速率等代谢参数与单次给药一致。可待因的最大血药浓度高于单次给药,但一个给药间隔期内的AUC值与单次给药相同。
磷酸可待因溶液[8]在人体内的半衰期为2.3h,达峰时间为0.5~1.5h。
Q.Y.YUE对中国人和白种人进行了一项磷酸可待因药物代谢(口服)对比研究[9]:可待因在中国人体内的平均Cmax和AUC值均比白种人高,而血浆清除率比白种人低,半衰期也比白种人长,在48h内通过尿液的累积排泄率与白种人接近。同时中国人体内可待因与C6G、吗啡、去甲吗啡的AUC值之比分别为1∶9、35∶1、4∶1,白种人体内的比值分别为1∶15、50∶1、6∶1。这一差异可能是由于中国人的葡萄糖醛酸化能力较弱引起的。
2.1.3 吗啡及其葡萄糖醛酸代谢物
超速代谢型人体内吗啡(包括其葡萄糖醛酸结合物)的平均血药浓度比正常代谢型人高50%[10],比弱代谢型人高45倍[11]。在超速代谢型人中,可待因发生O-脱甲基生成吗啡的比例超过15%;在正常代谢型人中,这一比例约为6%~10%;而在弱代谢型人中,可待因O-脱甲基的比例非常低(<1%)[12-14]。
弱代谢型人中吗啡和吗啡葡糖醛酸结合物的血药浓度、尿液回收率都非常低[8]11,15-16,几乎不产生止痛效果[17],对可待因的成瘾风险也低[18]。而在超速代谢型人中,可待因的成瘾和副作用风险较大,有报道称超速代谢型人在服用可待因后出现严重的阿片副作用[2,19]。
可待因被美国儿科协会和主流权威文章列为可随乳汁分泌的药物,曾有产妇(超速代谢型)使用可待因后导致其母乳喂养的新生儿死于吗啡中毒的情况发生[20]。
2.2 在相关疾病患者体内的药物代谢研究[21]
可待因需要在肝脏中代谢为吗啡才能发挥镇痛、止咳等作用,在肝功能不全患者中可待因药效减弱,如果重复用药还会导致可待因累积,因此对于肝功能不全患者应避免使用可待因药物。
可待因及代谢物C6G、吗啡等通过肾脏排泄,在中重度肾功能不全患者中可待因和代谢物的清除速率降低。而M6G等代谢物的累积有可能引发呼吸停止和发作性睡眠。对于超速代谢型人,肾功能不全引起的代谢物累积还可能导致中毒反应。为了避免代谢物累积产生的副作用,一般不建议对于肾功能不全患者使用可待因药物。
3 药物的相互作用研究
3.1 利福平[22]
利福平可以增加可待因的N-脱甲基和葡糖醛酸化,可待因的血药浓度降低、清除速率升高,而去甲可待因、去甲可待因葡糖醛酸和去甲吗啡的血药浓度升高。在异喹胍代谢能力高(CYP2D6活性高)的个体内,合用利福平后吗啡的血药浓度降低,药效减弱。
3.2 奎尼丁
奎尼丁是细胞色素P450的选择性抑制剂,在体外可以抑制可待因转化为吗啡[3]。可待因与奎尼丁合用后,血液中不能检测到吗啡和吗啡代谢物,可待因的O-脱甲基反应大幅降低[23]。
3.3 布洛芬
布洛芬与可待因口服合用,两者的药物代谢动力学无相互影响,而且两者在人体的半衰期接近[24-25],因此已被开发为复方制剂,但是近年有不少关于长期服用布洛芬-可待因复方制剂而导致低钾血症、肾小管酸中毒的案例[26-28]。
3.4 苯乙哌啶酮[29]
长期合用可待因和苯乙哌啶酮,可以影响大鼠的中枢神经系统,产生昏迷和成瘾,可能是由于药物对中枢神经的抑制作用增强。
3.5 其他
根据A Somogyi等人的研究,扑热息痛[30]、乙醇[31]、烟草[32]对可待因的药物代谢动力学均无明显影响。
4 可待因的毒性研究
通过大鼠或小鼠长期口腔给药研究,可待因并未产生靶器官毒性[33]和基因毒性[34]。高剂量可待因可使怀孕仓鼠和小鼠的肝重量增加,胎儿平均体重降低,并且仓鼠胎儿中出现了结构畸形。可待因产生生殖毒性的剂量比产生母体毒性的剂量低,在仓鼠中100mg/kg/天即可产生[35]。
5 可待因的药物依赖性
关于可待因产生依赖性的机理,一般认为与药物引起的神经适应性有关。连续用药2个月后,大鼠海马体中N-乙酰天冬氨酸、谷氨酸、胆碱、氨基乙磺酸的浓度均大幅降低[36]。对大鼠喂食含可待因的食物(0.5mg/g),仅喂食2天后大鼠体重减轻,并且喂食时间越长体重减轻越多,在停药后出现了腹泻、上睑下垂等症状[37]。
Zhen Cao等[38]对健康受试者进行了磷酸可待因的影响性研究,发现单次口服低剂量(1.0mg/kg)的磷酸可待因便可改变大脑不同分区的低频波动振幅和区域同质性,影响不同分区的大脑机能。
6 总结
可待因由法国化学家Robiquet于1832年首次发现,一个多世纪以来因其良好的止痛和止咳作用被广泛用于镇痛药和止咳药。通过大量的人体代谢研究和临床数据可知,可待因的人体代谢具有非常明显的个体差异和族群差异,尤其是发挥药理活性的O-脱甲基代谢,因此在不同代谢型人体中表现出不同的药理活性。临床用药时应根据患者代谢类型调整用法用量,避免长期用药产生依赖性,并避免对肝肾损伤患者、孕妇等特殊人群用药,以免产生药物蓄积毒性和生殖毒性。
