连锁与连锁不平衡联合作图解析烟草青枯病抗性遗传变异
2019-02-07赖瑞强李荣华夏岩石郭培国袁清华赵伟才张振臣
赖瑞强 李荣华 夏岩石 郭培国 袁清华 赵伟才 张振臣

摘 要:選用“粤烟98”ד118-3”构建的133份F7代单粒传重组自交系(RIL)群体和94份烟草种质组成的自然群体作为研究材料,运用253个SSR和293个InDel标记对这些材料进行基因分型,并针对烟草青枯病抗性开展连锁和连锁不平衡联合作图分析。结果显示,基于RIL群体的连锁分析在4个环境中共发现4个解释率介于12.00% ~ 30.10%之间的QTLs,利用自然群体的关联分析在3个环境中发现解释率在8.78%~11.42%之间的8个单倍型与抗病性密切相关,利用RIL群体和自然群体开展的连锁-连锁不平衡的整合作图分析发现8个解释率介于4.95%~6.93%之间的单倍型与抗病性密切相关。两种或以上分析方法均探测到与烟草青枯病抗性密切相关的单倍型有4个,分别为具正表型效应的Hap227和Hap368及负表型效应的Hap289和Hap370。所得结果表明连锁和连锁不平衡联合作图策略能较准确地发现与烟草青枯病抗性相关的QTLs,该结果可为烟草青枯病抗性相关研究及后续烟草青枯病抗性材料的辅助筛选提供参考。
关键词:烟草;青枯病抗性;连锁分析;关联分析;连锁和连锁不平衡联合作图
中图分类号:S572.03 文章编号:1007-5119(2019)06-0001-10 DOI:10.13496/j.issn.1007-5119.2019.06.001
Dissection of Genetic Variations for Tobacco Bacterial Wilt Resistance Based on Joint Linkage and Linkage Disequilibrium Mapping
LAI Ruiqiang1, LI Ronghua1, XIA Yanshi1, GUO Peiguo1*, YUAN Qinghua2,
ZHAO Weicai3, ZHANG Zhenchen2
(1. School of Life Sciences, Guangzhou University, Guangzhou 510006, China; 2. Guangdong Academy of Agricultural Sciences Institute of Crops, Guangzhou 510643, China; 3. Guangdong Tobacco Nanxiong Research Institute, Nanxiong, Guangdong 512400, China)
Abstract:A population with 133 F7 recombinant inbred lines (RIL) derived from the cross of “Yueyan 98” × “118-3” and a natural population with 94 tobacco accessions were used as materials, and joint linkage and linkage disequilibrium (LD) mapping analysis for tobacco bacterial wilt (TBW) resistance was carried out by using 253 SSR and 293 InDel markers. Four QTLs with 12%-30.10% of the explanation rate were detected by linkage analysis of the RIL population in four environments; and 8 haplotypes were found to be closely related to TBW resistance in three environments and explained 8.78%-11.42% of phenotypic variation by LD mapping with natural population. The integrated linkage-LD mapping of the RIL and natural populations was carried out and 8 haplotypes were detected to be closely associated with TBW resistance and explained 4.95%-6.93% of phenotypic variation. Four haplotypes were found to be associated with TBW resistance in at least two of the three analytical methods, two (Hap227 and Hap370) of them are with positive phenotypic effect, and the other two (Hap289, Hap368) are with negative effect. The results indicate that the strategy of joint linkage-LD mapping could accurately detect QTLs related to TBW resistance, and these haplotypes could provide reference for TBW resistance research and screening of TBW resistance materials.
Keywords:tobacco; bacterial wilt resistance; linkage analysis; association analysis; joint linkage and linkage disequilibrium mapping
烟草生长期间常常遭受到青枯病的危害,导致烟草减产和品质变劣[1],种植抗青枯病烟草品种是减少该病危害的有效途径,而挖掘出与青枯病抗性相关的分子标记用于辅助抗病材料的筛选,可加速青枯病抗病品种的选育进程。
选用家系群体开展连锁作图的QTL定位分析和利用自然群体开展的连锁不平衡作图分析是目前挖掘烟草青枯病抗病性状相关分子标记的两种有效方法。NISHI等[2]通过DH系群体构建遗传连锁图谱,首次对烟草青枯病抗性进行QTL定位,并发现1个与青枯病抗病性相关的QTL;随后,多位研究者通过构建遗传连锁图谱研究烟草青枯病抗性遗传,发现多个与青枯病抗病相关的QTLs[1,3-5];虽然这些利用连锁作图的QTL定位分析方法在研究青枯病抗性遗传方面已取得了一些成绩,但由于连锁分析是利用两个亲本所构建群体材料进行的研究,所涉及的仅限于同一基因座上的两个等位基因,研究的遗传背景较狭窄,且由于分离群体杂交或自交的次数有限,导致重组的次数较少,所以作图精度较低、分辨率较差[6-7]。而关联分析能克服连锁分析的这一缺点,它通过选用遗传背景差异大的种质即自然群体作为研究对象,不需构建作图群体,所花费的时间短、精力少[8],涵盖的遗传背景大,且利用了种质材料中多世代的重组事件,因此具有较高的分辨率[6]。目前,已有利用该方法对烟草青枯病抗性进行研究的报道[9-10]。但关联分析同样存在局限性,如对遗传多样性较低的群体其作图效果不如连锁分析,且关联分析不能确定QTL的具体位置及其加性效应;另外,关联分析不仅对稀有等位基因变异的发现能力较低,而且容易出现假阳性[11];而连锁分析能克服关联分析存在的这些不足[12]。所以,连锁分析和关联分析两种方法相结合的分析策略,可以扬长避短,提高目标性状QTL定位的準确性和精度[11,13]。基于连锁与连锁不平衡作图联合分析的方法已经在杨树[14]、水稻[15]和玉米[16]等植物中开展,且取得了不少成绩,但未见利用这种方法挖掘烟草青枯病抗性相关分子标记的报道。
据此,本研究选用“粤烟98”ד118-3”构建的133份单粒传重组自交系(RIL)和含有94份烟草种质的自然群体作材料,对这些材料的青枯病病情指数进行连锁与连锁不平衡联合作图分析。通过构建连锁图谱的QTL定位和连锁不平衡作图平行及整合作图分析,挖掘出与烟草青枯病抗性相关的分子标记,为分子标记辅助选择烟草青枯病抗性材料奠定理论基础。
1 材料与方法
1.1 试验材料
133份F7代单粒传重组自交系(RIL)由“粤烟98”(母本)ד118-3”(父本)衍生而来,其中“118-3”为高抗青枯病材料[10],而“粤烟98”为感病材料。94份烟草种质涵盖烤烟、雪茄烟、晒烟和白肋烟4种类型,来源地包括美国、日本、加拿大、索马里、澳大利亚、津巴布韦以及中国[10,17]。以上两个群体均由广东省烟草南雄科学研究所提供。
1.2 试验设计
2015年2月26日和2018年3月6日将供试材料的烟苗移栽至广东省烟草南雄科学研究所试验田,株距0.5 m,行距1.2 m,每份材料种植20株,田间管理按当地生产实际。以“岩烟97”和“红花大金元”分别作为抗病和感病对照。其中,RIL群体两年包括4个种植环境(E1~E4);自然群体两年包括3个种植环境(Ea~Ec)。此外,2015年的RIL群体(E1)和自然群体(Ea),2018年的RIL群体(E3)和自然群体(Eb)均种植于同一块试验田,环境变量一致,表型差异主要来源于材料的抗性差异,而环境代号不一致(如E1和Ea)仅为区分RIL群体和自然群体而设定。
1.3 青枯病病情调查
2015年5月4日和2018年5月6日按申莉莉[18]的方法使用致病小种1/生化型III的青枯病菌(Ralstonia solanacoarum)对处于旺长期的烟草进行茎部穿刺接种。于2015年6月6日和2018年6月5日进行病情等级调查,此时“红花大金元”的病情指数均接近100,调查标准参照GB/T 23222—2008 烟草病虫害分级及调查方法。并根据烟草病害等级计算病情指数(病指,Disease index,DI)[19],
用于后续的连锁/关联分析。
病指=∑(各级病株数×该病级值)/(调查总株数×最高级值)×100
1.4 DNA提取及基因分型
采取烟草新鲜叶片,使用李荣华等[20]改良的CTAB法进行DNA提取,并检测DNA的质量及浓度,随后将其稀释至20 ng/L,作为PCR扩增模板。
利用已报道的烟草SSR标记[21-25]及本实验室利用“118-3”和“粤烟98”进行RAD-seq(Restriction-site associated DNA sequence)开发的SSR和InDel标记,从中筛选出在“岩烟97”、“红花大金元”、“118-3”和“粤烟98”4份材料中扩增条带清晰易辨、特异性良好且具有多态性标记546个(其中SSR标记253个、InDel标记293个),用于群体基因分型。
SSR及InDel标记扩增的PCR反应体系及扩增程序详见文献[10]。采用6%非变性聚丙烯酰胺凝胶电泳和银染法[26]檢测PCR扩增产物,最后使用BenQ M800扫描仪扫描凝胶电泳图谱,通过GelBuddy软件[27]判读凝胶电泳图谱中的条带,根据条带的有无,确定等位变异位点,并构建1、0二元数据矩阵,完成群体基因分型。所用试剂均购自于生工生物工程(上海)股份有限公司。
1.5 数据分析
利用SPSS 19.0对烟草青枯病病指进行统计分析,包括平均值、标准差、变异系数及方差分析等;参考刘凯等[28]的方法计算广义遗传力。
1.6 遗传连锁图谱的构建和QTL定位
根据RIL群体基因分型数据,将与“118-3”相同的带型记为“A”,与“粤烟98”相同的带型记为“B”制成数据矩阵,并应用JoinMap 3.0软件构建烟草遗传连锁图谱,设置LOD≥3.0,重组率=0.4,选用Kosambi函数将重组率转换为图距单位(cM)并设置错误检测水平为1%。连锁群命名及顺序参考BINDLER等[21-22]的“LG+数字”命名方式和顺序;如果在图谱的构建过程中,少数连锁群因标记间遗传距离过大出现连锁群断点,但与BINDLER等[21-22]所建连锁群的标记相比仍属于同一连锁群时,则记为该连锁群A或B。
利用MapQTL 5.0软件进行QTL定位。首先,设置置换测验(Permutation Test)为1000,并在α=0.05的水平上预估每个连锁群的LOD阈值[29],结果以LOD≥2.6作为判断QTL存在的标准;然后,设置扫描各个连锁群的步移速度为1 cM,并进行区间作图(Interval mapping, IM),检测QTL的置信区间、加性效应及解释率。以LOD最高峰值的位置作为QTL的位置,而与之最近的左右标记分别称为左翼标记和右翼标记。
1.7 单倍型的构建及关联分析
分别利用Haploview软件[30]、SPAGeDi1.3d软件[31]和Structure 2.3软件进行单倍型构建、烟草材料间的亲缘系数分析和群体结构分析,而参数设置和数据处理方式均按赖瑞强等[10]的方法。随后,利用TASSEL软件中的混合线性模型(Mixed Linear Model,MLM)进行青枯病病情指数的关联分析。最后,采用BENJAMINI等[32]的方法,对p<0.01的显著关联单倍型进行控制错误发生率(False discovery rat,FDR)的假阳性验证;当FDR值也小于0.01时,表明对应的关联单倍型可靠性更高。
1.8 连锁与连锁不平衡联合作图及表型效应分析
依据LU等[33]连锁与连锁不平衡联合作图的分析方法。对连锁分析和关联分析的结果进行比较,开展连锁与连锁不平衡平行作图分析;整合作图分析则是利用共同的标记位点将133份RIL群体及亲本和94份自然群体整合为一个群体(含材料数228份),进行关联分析;其中,228份材料,前94个号(1~94)对应94份烟草种质的编号,后133个号(95~227)对应RIL群体的株系编号(1~133),而“粤烟98”的编号为228。
依据ZHANG等[34]的方法进行表型效应值计算。具体计算公式如下:
Ai=∑Xij/ni-∑Nk/nk
式中,Ai代表第i个单倍型的表型效应值,Xij为携带第i个单倍型的第j个材料表型的测定值,ni
则为具有第i个单倍型的材料数;∑Nk/nk代表所有材料表型测定值的平均值。
2 结 果
2.1 烟草青枯病病情指数
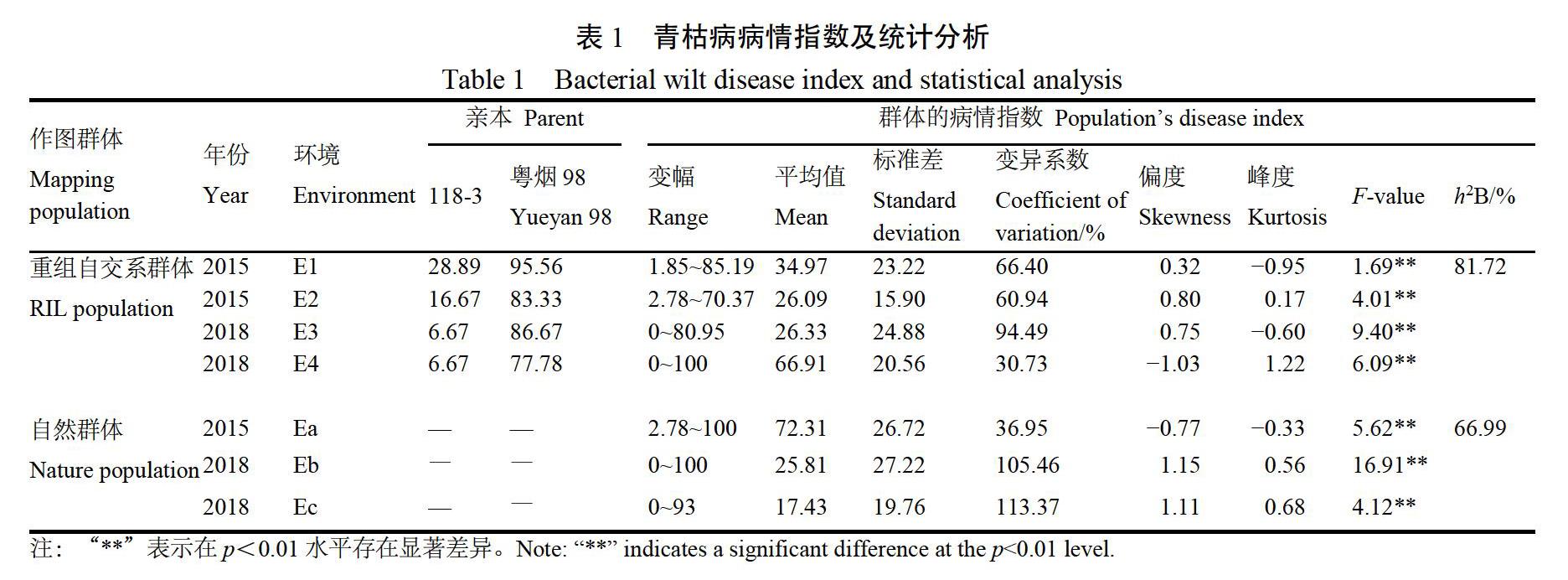
RIL群体和自然群体的青枯病病情指数鉴定结果如表1。RIL群体中,“118-3”的病情指数均明显低于“粤烟98”,表明亲本间的青枯病抗性差异大;其中,除了E4,E1~E3的病情指数偏度和峰度绝对值均小于1,符合正态分布的特点,且病情指数变异系数介于30.73%~94.49%,平均为63.14%,变异系数大,表明该性状的改良潜力大。而自然群体中,环境Ea中的病情指数变异系数为36.75%,其偏度和峰度的绝对值均小于1,符合正态分布的特征;而在环境Eb和Ec中变异系数和偏度均大于1,表明在这两个环境中材料表现出的抗性差异较大。
对同一环境下的RIL群体各株系间和自然群体各种质间的青枯病病情指数进行统计分析,发现其F值介于1.69~16.91之间,且均达到极显著水平,表明在同一环境中两个作图群体的各材料间的青枯病抗性存在显著差异;此外,两个群体的青枯病病情指数的广义遗传率均超过60%,表明基因对该性状也存在較大的影响。因此,烟草材料间的青枯病病情指数差异明显,有利于进行QTL定位/关联分析。
2.2 连锁图谱的构建及青枯病抗性的QTL定位
利用546个标记对RIL群体进行基因分型,共发现1043个变异位点;利用这些变异位点数据进行连锁作图分析,构建了一张包括24个连锁群的遗传图谱。该图谱包含463个标记,占总标记数的84.62%,覆盖基因组总长度为1230.72 cM,两标记间的平均遗传距离为2.66 cM。
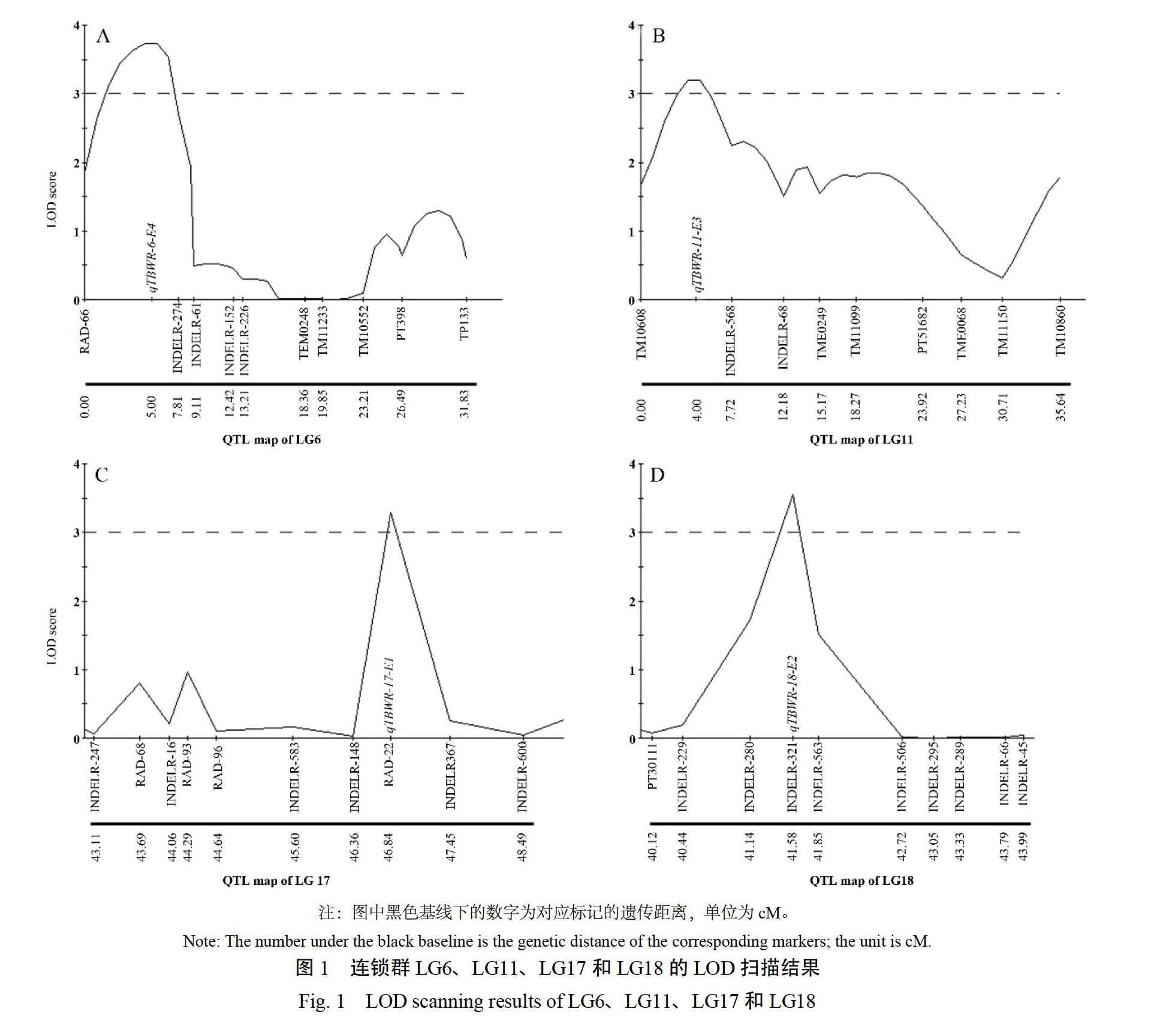
基于该遗传图谱,对RIL群体2015年和2018年的青枯病病情指数进行QTL定位。两年间共发现qTBWR-6-E4、qTBWR-11-E3、qTBWR-17-E1和qTBWR-18-E2等4个QTLs(图1,表2),这些QTLs分别位于LG6、LG11、LG17和LG18上,贡献率介于12%~30.10%之间;其中qTBWR-17-E1的加性效应为负值,表明此效应来自抗性亲本“118-3”,可能关联到抗病的等位基因;其余3个QTL的加性效应为正值,表明此加性效应来源于感病亲本“粤烟98”,可能关联到与青枯病感病相关的等位基因。
2.3 连锁不平衡分析和单倍型构建
R2值是衡量两个位点连锁不平衡程度的重要指标,与关联分析的效力有关[35]。因此,对利用546个分子标记对94份自然群体进行基因分型获得的1503个变异位点两两之间(共1128753个组合)的R2值进行计算,发现它们之间的R2值范围为0~1,具较强的连锁不平衡的组合(即R2≥0.216)[36]占总组合的7.57%,R2>0.8的组合占总组合的2.14%,表明该群体存在一定的连锁不平衡,可进行关联分析[37]。当R2值大于0.8时表明两个位点存在强连锁
不平衡,可被归入同一个单倍型中[38]。因此,在R2>0.8的条件下,对自然群体基因分型中发现的1503个变异位点进行单倍型分析,共构建了376个单倍型,且所有单倍型的基因频率均大于5%。
2.4 群体结构分析和关联分析
运用376个单倍型对94份烟草种质进行群体结构分析,发现当最佳类群数K=2时,△K值最大,因此将其分为两个类群;这一结果表明应用于本研究的材料群体结构简单,而简单的群体结构有利于关联分析[40]。
据此,本研究结合群体结构Q值和材料间的亲缘系数值,基于376个单倍型,对2015年和2018年统计的青枯病病情指数进行关联分析,并对显著关联(p<0.01)的单倍型进行假阳性检验(FDR< 0.01),确定出8个单倍型与青枯病抗性密切相关(表3);其中,2015年Ea环境有Hap360、Hap362和Hap370等3个单倍型与青枯病抗性密切相关,表型变异解释率分别为9.27%、11.42%和11.42%;2018年在环境Eb中有3个单倍型即Hap126、Hap227和Hap289与青枯病抗病性显著相关,表型变异解释率分别为8.78%、8.78%和11.33%;在2018年Ec环境中有2个单倍型Hap28和Hap59与青枯病抗病性显著相关,表型变异解释率分别为8.27%和10.21%。
2.5 连锁与连锁不平衡平行作图
对连锁分析和关联分析的结果进行比较,发现在94份材料的关联分析中检测到的8个单倍型,其中有2个单倍型即Hap227和Hap370中含有的分子标记与连锁分析检测到的QTL位点处于同一个连锁群上。其中,Hap227含有的分子标记INDELR-229与qTBWR-18-E2在LG18上峰值的距离为1.14 cM(图1 D);Hap370含有的分子标记RAD-68与qTBWR-17-E1在LG17上峰值的距离为3.15 cM(图1 C)。但连锁分析中探测到的另两个QTLs(qTBWR-6-E4和qTBWR-11-E3)未发现与关联分析存在联系,关联分析中获得的其他6个单倍型亦未发现与连锁分析探测到的QTL存在关联。
2.6 228份烟草材料的群体结构及整合作图
利用546个标记对整合群体进行基因分型,确定出1399个等位基因频率均大于0.05的变异位点,并在R2>0.8的条件下构建了573个单倍型,利用单倍型进行群体结构分析,将其分为两个类群。
对2015年和2018年的病情指数进行关联分析,并经FDR验证,共发现到8个单倍型与烟草青枯病抗性密切相关(表4)。其中,2015年共检测到Hap9、Hap15、Hap250、Hap261、Hap370和Hap368等6个单倍型与青枯病抗性紧密相关,表型变异解释率介于5.03%~6.50%之间;而2018年仅检测到Hap73和Hap289等2个单倍型与青枯病抗性紧密相关,表型变异解释率分别为4.95%和6.93%。
整合作图中探测到的Hap289和Hap370在利用94份烟草材料进行的关联分析中亦被探测到,单倍型Hap368中涉及的分子标记RAD-66是连锁分析中QTL qTBWR-6-E4的左翼标记。因此,单倍型Hap289、Hap370和Hap368适用的材料范围可能较广,可靠性也较高,可利用其进行抗性材料的筛选。
2.7 青枯病抗病相关单倍型的表型效应
在连锁分析、关联分析和整合作图分析中,有4个单倍型(Hap227、Hap289、Hap370和Hap368)在两种及以上分析方法均发现与抗病密切相关,表明这些单倍型用于评价青枯病抗病性将具有较高的准确性。因此,对它们进行表型效应分析(表5),发现Hap289和Hap370表现为减效效应;而Hap227和Hap368表现为增效效应。
3 讨 论
本研究利用建立的遗传图谱进行青枯病抗性QTL定位,检测到4个解释率在12%~30.1%之间的QTLs,表明这些可能为主效基因位点,与NISHI等[2]、MATSUDA等[40]和MATSUDA[41]研究发现烟草青枯病抗性有主效基因影响的结论一致。此外,所发现的QTL qTBWR-6-E4的位置与SSR标记TP398和TP133的遗传距离分别为21.49 cM和26.83 cM(图1),与范江[42]研究发现青枯病抗病品种“R8”中抗病基因与标记TP398和TP133的遗传距离分别为24.49 cM和26.51 cM的结果基本一致,猜测qTBWR-6-E4可能为“R8”的抗性基因;该QTL左右两翼邻近标记RAD-66和INDELR-27与QTL峰值点的距离均小于6 cM,表明连锁得更加紧密,因此利用这两个分子标记对烟草材料进行抗病性评价更具优势。值得一提的还有,本研究发现4个QTL的临近标记多为利用“粤烟98”和“118-3”这两个亲本基于RAD-seq测序开发的InDel标记;
由于InDel标记在基因组中分布广泛,扩增产物的稳定性和分辨率均较SSR标记好[43],而以往的报道中利用AFLP、RAPD、SSR等标记对烟草青枯病抗性进行QTL定位[1-3,5,44],而未见有利用InDel标记开展烟草遗传图谱的构建和青枯病抗性QTL定位工作;因此本研究丰富了烟草青枯病研究的标记类型,有利于后续对烟草材料进行更精确的评价。
连锁分析的QTL定位主要利用两个亲本的遗传背景,由此获得的分子标记适用的材料范围有待考量[10];而利用自然群体作材料开展的基于连锁不平衡的关联分析却能克服上述缺点,挖掘出的分子标记将可能具有较为广泛的适用性。目前在烟草青枯病抗病性关联分析的研究报道较少,仅见2篇我们前期分别利用37个MFLP标记和236个SSR標记对烟草青枯病抗性进行关联分析[9-10]。本研究在前期研究工作基础上,运用253个SSR标记和293个InDel标记共546个标记开展青枯病抗性的关联分析,增加了标记数目和标记的类型,且使用单倍型进行基于连锁不平衡的关联分析,发现了解释率介于8.27%~11.42%之间的8个单倍型(含16个变异位点)与青枯病抗病性相关,所发现到的单倍型数多于赖瑞强等[10]检测到的7个单倍型数,涉及的变异位点数也远多于吴超等[9]利用变异位点进行关联分析发现到6个与青枯病抗性相关的MFLP变异位点;而值得一提的是本研究所发现单倍型Hap59,所含的变异位点即TM10247.125/TM10247.130在赖瑞强等[10]研究中也被关联到与烟草青枯病抗性相关,表明本研究的关联结果可靠性高,可为烟草青枯病抗病材料的分子标记辅助筛选提供参考。
虽然关联分析能克服连锁分析遗传背景狭窄的缺点,但也存在局限性,如关联分析不能检测QTL的加性效应,且对稀有等位基因的检测效能低[11];反之,连锁分析却不具有上述缺点[12]。因此,连锁分析和关联分析两种分析方法各有优劣,通过结合这两种方法,扬长避短进行相互验证有利于提高QTL定位的准确性[10]。在本研究开展的连锁与连锁不平衡联合作图的平行作图中,发现与抗病性相关的单倍型Hap227和Hap370分别与坐落在连锁群LG18的qTBWR-18-E2和LG17上qTBWR-17-E1峰值之间的遗传距离均小于4 cM;而刘凯等[28]报道当关联位点与QTL在连锁群上的遗传距离小于6 cM时,表明对应区段存在目标性状的相关基因;因此本研究发现到的两个区段,可能存在与烟草青枯病抗性相关的基因。但本研究也存在一些QTL和关联位点没有相互验证到的情况,这可能是两个群体的遗传背景差异造成,该结果与LU等[33]认为关联分析中检测到的一些标记未被连锁分析发现,是由于群体遗传背景差异造成的结论一致。而在连锁与连锁不平衡联合作图的整合作图中,共发现到8个单倍型与烟草青枯病抗性相关,其中Hap289和Hap370在利用种质材料的关联分析中亦被发现,而Hap368含有RAD-66标记位点,为本研究连锁分析探测到的QTL(qTBWR-6-E4)侧翼标记,且与范江[42]发现到的青枯病抗性基因位置基本一致。这些说明了本研究结果的具有较高的准确性。
4 结 论
本研究首次利用连锁与连锁不平衡联合作图策略对烟草青枯病抗性进行研究,探测到4个在两种及以上分析方法均发现与抗病密切相关单倍型。该结果可为烟草青枯病抗性的相关研究及后续烟草青枯病抗性材料的辅助筛选提供参考。
参考文献
[1] LAN T, ZHENG S, YANG L, et al. Mapping of quantitative trait loci conferring resistance to bacterial wilt in tobacco (Nicotiana tabacum L.)[J]. Plant Breeding, 2015, 133(5): 672-677.
[2] NISHI T, TAJIMA T, NOGUCHI S, et al. Identification of DNA markers of tobacco linked to bacterial wilt resistance[J]. Theoretical and Applied Genetics, 2003, 106(4): 765-770.
[3] QIAN Y, WANG X, WANG D, et al. The detection of QTLs controlling bacterial wilt resistance in tobacco (N. tabacum, L.)[J]. Euphytica, 2013, 192(2): 259-266.
[4] DRAKE-STOWE K, BAKAHER N, GOEPFERT S, et al. Multiple disease resistance loci affect soilborne disease resistance in tobacco (Nicotiana tabacum)[J]. Phytopathology, 2017, 107(9): 1055-1061.
[5] 袁清华,马柱文,张振臣,等. 烟草品种大叶密合抗青枯病相关QTL的检测[J]. 中国烟草学报,2018,24(3):77-81.
YUAN Q H, MA Z W, ZHANG Z C, et al. Detection of QTL associated with tobacco bacterial wilt resistance in cultivar Dayemihe[J]. Acta Tabacaria Sinica, 2018, 24(3): 77-81.
[6] FLINT-GARCIA S A, THUILLET A C, YU J, et al. Maize association population: a high-resolution platform for quantitative trait locus dissection[J]. Plant Journal for Cell & Molecular Biology, 2005, 44(6): 1054-1064.
[7] SALVI S, TUBEROSA R. To clone or not to clone plant QTLs: present and future challenges[J]. Trends in Plant Science, 2005, 10(6): 297-304.
[8] ERSOZ E S, YU J, BUCKLER E S. Applications of linkage disequilibrium and association mapping in crop plants[M]//Genomics-assisted crop improvement. Dordrecht: Springer Netherlands, 2007: 97-119.
[9] 吳超,夏岩石,李荣华,等. 烟草青枯病抗性与分子标记的关联分析[J]. 烟草科技,2015(10):1-12.
WU C, XIA Y S, LI R H, et al. Association analysis of tobacco bacterial wilt resistance with molecular markers[J]. Tobacco Science & Technology, 2015(10): 1-12.
[10] 赖瑞强,李荣华,夏岩石,等. 烟草种质的SSR标记遗传多样性及青枯病抗性的关联分析[J]. 中国烟草学报,2018,24(6):67-77.
LAI R Q, LI R H, XIA Y S, et al. SSR marker-based genetic diversity analysis of tobacco germplasm and association analysis with resistance to bacterial wilt[J]. Acta Tabacaria Sinica, 2018, 24(6): 67-77.
[11] MACKAY I, POWELL W. Methods for linkage disequilibrium mapping in crops[J]. Trends in Plant Science, 2007, 12(2): 57-63.
[12] COLLARD BCY , JAHUFER MZZ , BROUWER J B, et al. An introduction to markers, quantitative trait loci (QTL) mapping and marker-assisted selection for crop improvement: the basic concepts[J]. Euphytica, 2005, 142(1-2): 169-196.
[13] MYLES S, PEIFFER J, BROWN P, et al. Author information. association mapping: critical considerations shift from genotyping to experimental design[J]. Plant Cell, 2009, 21(8): 2194-2202.
[14] DU Q, GONG C, WANG Q, et al. Genetic architecture of growth traits in populus revealed by integrated quantitative trait locus (QTL) analysis and association studies[J]. New Phytologist, 2015, 209(3): 1067-1082.
[15] LOU Q, CHEN L, MEI H, et al. Quantitative trait locus mapping of deep rooting by linkage and association analysis in rice[J]. Journal of Experimental Botany, 2015, 66(15): 4749-4757.
[16] ZHANG C, ZHOU Z, YONG H, et al. Analysis of the genetic architecture of maize ear and grain morphological traits by combined linkage and association mapping[J]. Theoretical and Applied Genetics, 2017, 130(5): 1011-1029.
[17] 刘文杰,李荣华,夏岩石,等. 利用荧光SSR和MFLP标记技术分析烟草核心种质的遗传多样性[J]. 分子植物育种,2016,14(10):2869-2881.
LIU W J, LI R H, XIA Y S, et al. Analysis of genetic diversity for core tobacco germplasm by using fluorescent SSR and fluorescent MFLP marker techniques[J]. Molecular Plant Breeding, 2016, 14(10): 2869-2881.
[18] 申莉莉. 煙草突变体筛选与鉴定方法篇:2.烟草抗主要病虫害突变体的筛选与鉴定[J]. 中国烟草科学,2012,33(2):102-104.
SHEN L L. Screening and identification methods of tobacco mutants: 2. screening and identification of tobacco resistant mutants to main pests and diseases[J]. Chinese Tobacco Science, 2012, 33(2): 102-104.
[19] 高加明,王志德,张兴伟,等. 香料烟青枯病抗性基因的遗传分析[J]. 中国烟草科学,2010,31(1):1-4.
GAO J M, WANG Z D, ZHANG X W, et al. Genetic analysis on resistance to bacterial wilt in oriental tobacco[J]. Chinese Tobacco Science, 2010, 31(1): 1-4.
[20] 李荣华,夏岩石,刘顺枝,等. 改进的CTAB提取植物DNA方法[J]. 实验室研究与探索,2009,28(9):14-16.
LI R H, XIA Y S, LIU S Z, et al. CTAB-improved method of DNA extraction in plant[J]. Research and Exploration in Laboratory, 2009, 28(9): 14-16.
[21] BINDLER G, PLIESKE J, BAKAHER, N, et al. A high density genetic map of tobacco (Nicotiana tabacum L.) obtained from large scale microsatellite marker development[J]. Theoretical & Applied Genetics, 2011, 123(2): 219-230.
[22] BINDLER G, VAN Der HOEVEN R, GUNDUZ I, et al. A microsatellite marker based linkage map of tobacco[J]. Theoretical & Applied Genetics, 2007, 114(2): 341-349.
[23] TONG Z, YANG Z, CHEN X, et al. Large-scale development of microsatellite markers in Nicotiana tabacum and construction of a genetic map of flue-cured tobacco[J]. Plant Breeding, 2012, 131(5): 674-680.
[24] 范江,劉勇,童治军,等. 烤烟品种‘Oxford207青枯病抗性的遗传分析与分子标记初选[J]. 中国农学通报,2013,29(34):50-55.
FAN J, LIU Y, TONG Z J, et al. Inheritance of resistance to Ralstonia solanacearum in flue-cured tobacco ‘Oxford07 and molecular marker screen[J]. Chinese Agricultural Science Bulletin, 2013, 29(34): 50-55.
[25] 李海洋,李荣华,夏岩石,等. 基于RAD-seq数据开发烟草多态性SSR标记[J]. 中国烟草科学,2018,39(1):1-9.
LI H Y, LI R H, XIA Y S, et al. Development of polymorphic SSR markers in tobacco based on RAD sequencing[J]. Chinese Tobacco Science, 2018, 39(1): 1-9.
[26] 郭培国,刘文杰,李海洋,等. 一种快速有效检测SSR标记的非变性聚丙烯酰胺凝胶的银染方法[J]. 广州大学学报(自然科学版),2016,15(4):8-12,49.
GUO P G, LIU W J, LI H Y, et al. A rapid and effective method of silver staining for detecting SSR markers in nondenaturing polyacrylamide gels[J]. Journal of Guangzhou University(Natural Science Edition), 2016, 15(4): 8-12, 49.
[27] ZERR T, HENIKOFF S. Automated band mapping in electrophoretic gel image using background information[J]. Nucleic Acids Research, 2005, 33(9): 2806-2812.
[28] 刘凯,邓志英,张莹,等. 小麦茎秆断裂强度相关性状QTL的连锁和关联分析[J]. 作物学报,2017,43(4):483-495.
LIU K, DENG Z Y, ZHANG Y, et al. Linkage analysis and genome-wide association study of QTLs controlling stem-breaking-strength-related traits in wheat[J]. Acta Agronomica Sinica, 2017, 43(4): 483-495.
[29] CHURCHILL G A, DOERGE R W. Empirical threshold values for quantitative trait mapping [J]. Genetics, 1994, 138(3): 963-971.
[30] BARRETT J C, FRY B, MALLER J, et al. Haploview: analysis and visualization of LD and haplotype maps[J]. Bioinformatics, 2005, 21(2): 263-5.
[31] YU J, PRESSOIR G, BRIGGS W H, et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness[J]. Nature Genetics, 2006, 38(2): 203-208.
[32] BENJAMINI Y H , HOCHBERG Y. Controlling the false discovery rate-a practical and powerful approach to multiple testing[J]. Journal of the Royal Statistical Society. Series B: Methodological, 1995, 57: 289-300.
[33] LU Y, ZHANG S, SHAH T, et al. Joint linkage–linkage disequilibrium mapping is a powerful approach to detecting quantitative trait loci underlying drought tolerance in maize[J]. Proc Natl Acad Sci USA, 2010, 107(45): 19585-19590.
[34] ZHANG T, QIAN N, ZHU X, et al. Variations and transmission of QTL alleles for yield and fiber qualities in upland cotton cultivars developed in China[J]. PLoS One, 2013, 8(2): e57220.
[35] 何云刚,金力,黄薇. 单核苷酸多态性与连锁不平衡研究进展[J]. 基础医学与临床,2004,24(5):487-490.
HE Y G, JIN L, HUANG W. Advance in the research of single nucleotide polymorphism and linkage disequilibrium[J]. Basic Medical Sciences and Clinics, 2004, 24(5): 487-490.
[36] 李英慧,袁翠平,张辰,等. 基于大豆胞囊线虫病抗性候选基因的SNP位点遗传变异分析[J]. 遗传,2009,31(12):1259-1264.
LI Y H, YUAN C P, ZHANG C, et al. Genetic variation of SNP loci based on candidate gene for resistance to soybean cyst nematode[J]. Hereditas, 2009, 31(12): 1259-1264.
[37] 谭贤杰,吴子恺,程伟东,等. 关联分析及其在植物遗传学研究中的应用[J]. 植物学报,2011,46(1):108-118.
TAN X J, WU Z K, CHENG W D, et al. Association analysis and its application in plant genetic research[J]. Bulletin of Botany, 2011, 46(1): 108-118.
[38] PHILLIPS M S, LAWRENCE R, SACHIDANANDAM R, et al. Chromosome-wide distribution of haplotype blocks and the role of recombination hot spots[J]. Nature Genetics, 2003, 33(3): 382-387.
[39] 王榮焕,王天宇,黎裕. 关联分析在作物种质资源分子评价中的应用[J]. 植物遗传资源学报,2007,8(3):366-372.
WANG R H, WANG T Y, LI Y. Application of association analysis in molecular evaluation of crop germplasm resources[J]. Journal of Plant Genetic Resources, 2007, 8(3): 366-372.
[40] MATSUDA T, OHASHI Y. Inheritance of resistance to bacterial wilt resistant varieties in tobacco[J]. Jap. Tour. Breeding, 1973, 23(4): 175-180.
[41] MATSUDA T. Fundamental studies on the breeding of bacterial wilt resistant varieties in tobacco[J]. Bulletin of the Utsunomiya Tobacco Experiment Station, 1997, 15: 1-95.
[42] 范江. 烟草种质青枯病抗性鉴定与分子标记筛选[D]. 长沙:湖南农业大学,2012.
FAN J. Tobacco germplasm bacterial wilt resistance identification and molecular marker screening[D]. Changsha: Hunan Agricultural University, 2012.
[43] 冯芳君,罗利军,李荧,等. 水稻InDel和SSR标记多态性的比较分析[J]. 分子植物育种,2005,3(5):725-730.
FENG F J, LUO L J, LI Y, et al. Comparative analysis of polymorphism of InDel and SSR markers in rice[J]. Molecular Plant Breeding, 2005, 3(5): 725-730.
[44] 杨友才,周清明,朱列书. 烟草青枯病抗性基因的遗传分析及RAPD标记[J]. 中国烟草学报,2006,12(2):38-42.
YANG Y C, ZHOU Q M, ZHU L S. Heredity and RAPD markers analysis of resistance gene to tobacco bacterial wilt[J]. Acta Tabacaria Sinica, 2006, 12(2): 38-42.
