尼美舒利原料药含量的2种测定方法的比较
2019-01-21朱跃芳
罗 诚,陈 莉,朱跃芳
尼美舒利是一种非甾体抗炎药,能选择性地抑制环氧化酶COX2,在发挥解热镇痛消炎作用的同时,减少了消化性溃疡等非甾体抗炎药物常见的不良反应。临床上常用于慢性关节炎、术后或急性创伤后疼痛、急性上呼吸道感染等疾病的治疗[1,2]。 尼美舒利分子中含磺酰胺结构,具有弱酸性,《中国药典》2015年版二部采用电位滴定法,以氢氧化钠滴定液测定尼美舒利原料药的含量[3]。其他文献中,尼美舒利制剂的含量测定以及血药浓度监测常采用高效液相色谱法、紫外分光光度法、薄层扫描法等[4-6],少有以高效液相色谱法测定尼美舒利原料药含量的报道。
笔者采用高效液相色谱法测定尼美舒利原料药的含量,并与《中国药典》2015年版二部中的电位滴定法进行比较,现报告如下。
1 仪器与试药
Agilent 1260高效液相色谱仪(美国安捷伦公司);Agilent TC-C18色谱柱(250.0mm×4.6mm,5μm)、Inertsil ODS-3 色谱柱(250.0 mm×4.6 mm,5 μm)、资生堂 CAPCELL PAK C18色谱柱(250.0 mm×4.6 mm,5 μm)、Waters XTerra C18色谱柱(250.0 mm×4.6 mm,5 μm);916 Ti-Touch 自动电位滴定仪(瑞士万通公司);888 Titrino自动电位滴定仪(瑞士万通公司);ZDJ-3D全自动电位滴定仪 (北京先驱星科技发展有限公司);848 Titrino plus自动电位滴定仪(瑞士万通公司);AG-135电子分析天平(梅特勒-托利多)。
尼美舒利对照品 (批号100555-201202,含量100.0%,中国食品药品检定研究院);尼美舒利原料药(批号 141182、141183,以下分别用 1#、2# 代替,天津药物研究院药业);丙酮(批号200808、201012,分析纯,中国医药集团上海化学试剂公司)、丙酮(批号201205、201307,色谱纯,天津市化学试剂研究所),磷酸、氨水均为分析纯,水为超纯水。
2 方法与结果
2.1 高效液相色谱法
2.1.1 色谱条件 采用Agilent TC-C18色谱柱(250 mm×4.6 mm,5 μm); 流动相为 0.1%磷酸溶液(用氨水调 pH 至 7.0)-乙腈 (60∶40); 流速 1.0 ml/min,检测波长 393 nm,进样量 10 μl。
2.1.2 溶液的配制 对照品溶液的配制:精密称取经105℃干燥2 h的尼美舒利对照品约20 mg,置100 ml量瓶中,加流动相溶解并稀释至刻度,得对照品储备液,精密量取对照品储备液10 ml置20 ml量瓶中,加流动相稀释至刻度,摇匀,得到浓度约为100 μg/ml的对照品溶液。同法制备供试品溶液。
2.1.3 专属性试验 取2.1.2项下制备的对照品溶液和供试品溶液各10 μl,注入高效液相色谱仪,记录色谱图和光谱图。图1~4表明,供试品和对照品在该色谱条件下出峰时间吻合,峰型良好,光谱图一致,说明该方法专属性良好。
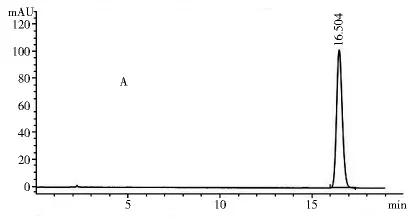
图1 对照品色谱图
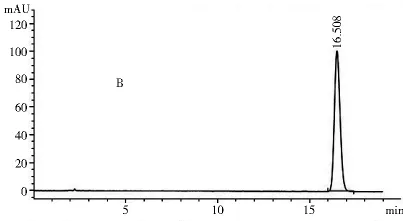
图2 供试品色谱图
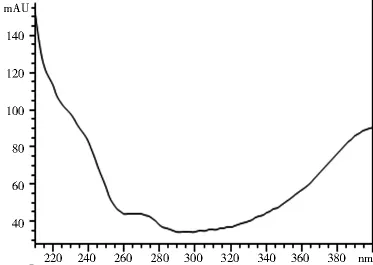
图3 对照品光谱图
2.1.4 线性范围考察 精密量取2.1.2项下的对照品储备溶液适量,用流动相稀释成浓度为10、40、80、120、160、200 μg/ml的梯度对照品溶液。 精密量取各梯度对照品溶液10 μl,注入液相色谱仪,以尼美舒利浓度为横坐标(X),色谱峰面积为纵坐标(Y)进行回归分析,得到回归方程为:Y=21.109X-1.583,r=1.000,说明尼美舒利在 10.00~200 μg/ml的浓度范围内线性关系良好。
2.1.5 精密度试验 取同一供试品溶液(批号1#),按2.1.1项下的方法连续进样6次,记录色谱图,色谱峰面积RSD(n=6)为0.2%,说明仪器精密度良好。
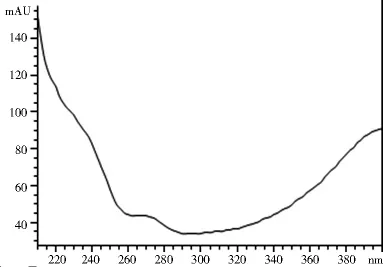
图4 供试品光谱图
2.1.6 重复性试验 取同一批尼美舒利原料药(批号1#),按2.1.2项下的方法制备供试品溶液共6份,进行平行测定,结果6次测定的平均含量为99.73%,RSD(n=6)为 0.2%,说明该方法重复性良好。
2.1.7 稳定性试验 取同一供试品溶液,在室温下放置 0、2、4、6、12、24 h 后进样, 记录色谱峰面积,RSD(n=6)为 0.4%,说明供试品溶液在 24 h内稳定。
2.1.8 加样回收率试验 将供试品(批号1#,含量99.73%)称样量减半,共称取9份,分成3组,每组分别加入相当于供试品80%、100%、120%的尼美舒利对照品,配制成供试品溶液。按2.1.1的条件进行测定,计算加样回收率,结果9份供试品加样回收率为98.61%~100.61%,平均加样回收率为99.52%,RSD(n=9)为 0.7%。 见表 1。
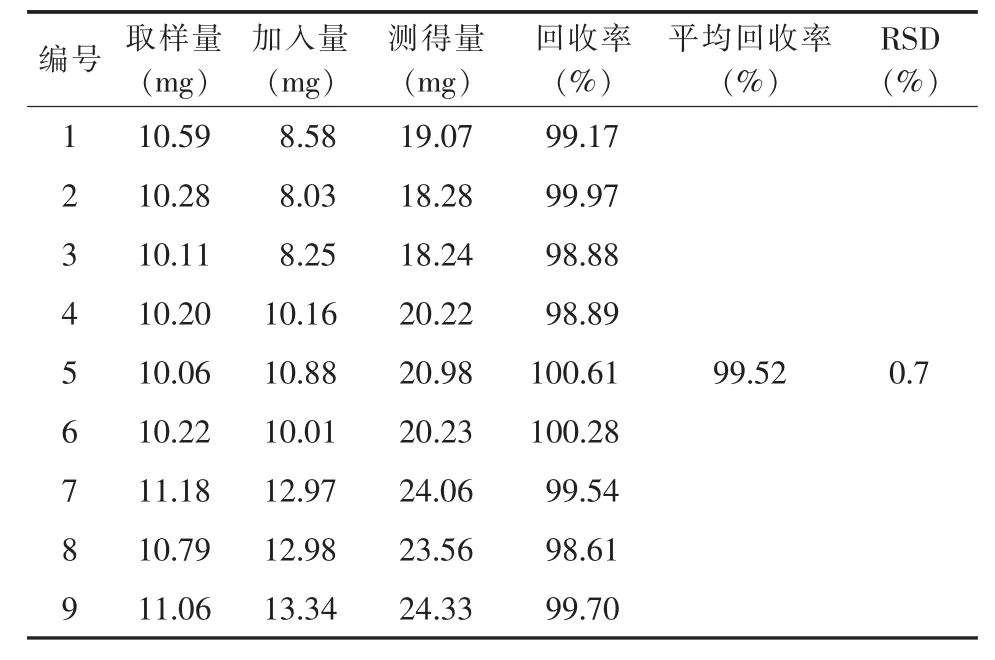
表1 HPLC法加样回收率测定结果
2.1.9 方法耐用性考察 采用同一批尼美舒利原料药(批号 1#),按 2.1.2 项下的方法制备供试品溶液,分别使用Agilent TC-C18色谱柱 (250 mm×4.6 mm,5 μm)、Inertsil ODS-3 色谱柱 (250 mm×4.6 mm,5 μm)、 资生堂 CAPCELL PAK C18色谱柱(250 mm×4.6 mm,5 μm)、Waters XTerra C18色谱柱(250 mm×4.6 mm,5 μm)测定供试品含量,结果含量分别为 99.73%、100.12%、99.79%、99.68%,RSD 为0.2%。将上述供试品溶液,采用 Agilent TC-C18色谱柱(250 mm×4.6 mm,5 μm), 在柱温为 25 ℃、30 ℃、35℃、40℃下分别进样,计算含量,结果分别为99.80%、99.92%、100.06%、99.71%,RSD 为 0.2%。改变流动相 pH 值为 6.8、7.0、7.2 测定供试品含量,每个pH值下各平行测定两次,结果主峰拖尾因子均<2, 含量分别为 99.87%、99.75%、99.89%、100.00%、100.03%、99.51%,RSD 为 0.2%。 改变流动相比例为(55∶45)、(60∶40)、(65∶35)测定供试品含量,每个条件下各平行测定两次,结果主峰与各杂质峰达到基线分离,拖尾因子均<2,含量分别为 99.96%、99.77%、99.90%、100.15%、99.74%、100.04%,RSD 为 0.2%。说明该方法耐用性良好。
2.2 电位滴定法
2.2.1 电位滴定参数 样品滴定参数:采用DET动态滴定模式,等当点判据标准30 mV,信号漂移20 mV/min,等当点后体积 1.0 ml。空白滴定参数:采用MET等量滴定模式,等当点判据标准5 mV,滴定液增量 0.005 ml/min,信号漂移 10 mV/min,等当点后体积 0.5 ml。
2.2.2 测定方法[3]精密称取尼美舒利原料药约0.25 g,加中性丙酮(对酚酞指示液显中性)40 ml使其溶解,加水20 ml,照电位滴定法(《中国药典》2015年版四部通则 0701),用氢氧化钠滴定液(0.1 mol/L)滴定,并将结果用空白试验校正。每1 ml氢氧化钠滴定液(0.1 mol/L)相当于 30.83 mg 的 C13H12N2O5S。
2.2.3 重复性试验 取同一批尼美舒利原料药(批号 1#),精密称取 6 份,按 2.2.2 的方法在 916 Ti-Touch自动电位滴定仪上测定,得到平均含量为100.04%,RSD(n=6)为 0.5%。
2.2.4 加样回收率试验 取已知含量的同一批尼美舒利原料药(批号 1#,含量 100.04%),按 2.2.2 项下的方法称取供试品6份,供试品取样量减半,加入相当于供试品100%的对照品后,在916 Ti-Touch自动电位滴定仪上测定,计算回收率,结果见表2。
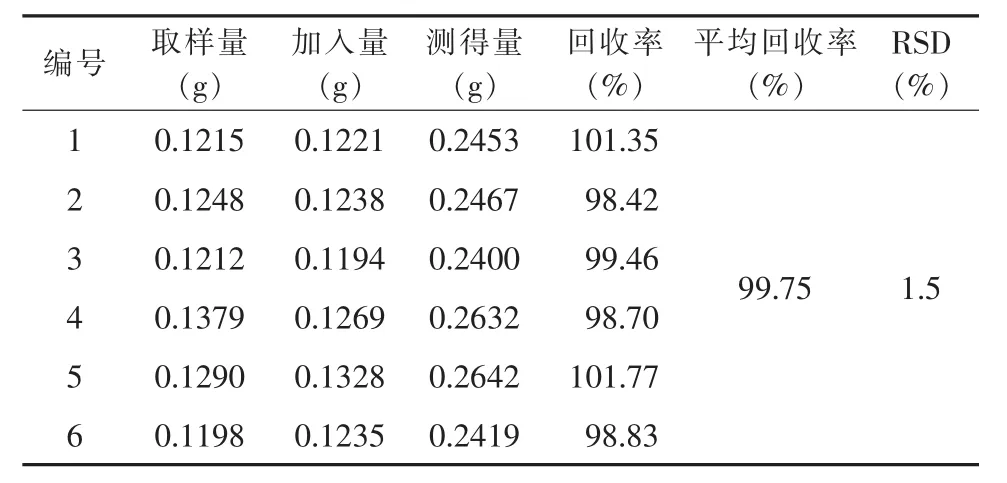
表2 电位滴定法加样回收率测定结果
2.2.5 中性丙酮的考察 在调制中性丙酮(对酚酞指示液显中性)时发现,酚酞在丙酮中显色不明显,终点难以判断。若滴加的氢氧化钠滴定液过量,多余的氢氧化钠会与尼美舒利反应造成结果偏低。该次实验考察了中性丙酮(显微红)、中性丙酮(稍显微红)及不调中性丙酮对结果的影响,结果见表3。可以看出,当电位滴定参数设置合理时,由于空白已扣除,丙酮是否调中性对结果几乎无影响,但调过头会导致结果偏低。由于空白滴定突跃不明显,应适当将“等当点后体积”适当设大,以防出现伪终点。
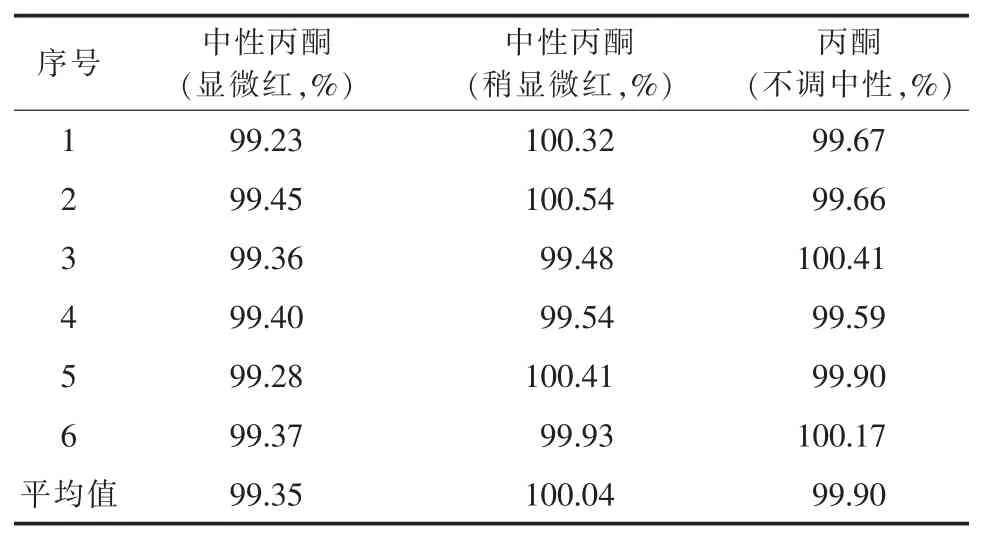
表3 中性丙酮考察结果
2.2.6 方法耐用性考察 采用同一批尼美舒利原料药(批号 1#),按 2.2.2 项下的方法制备供试品溶液,分别使用916 Ti-Touch自动电位滴定仪(瑞士万通公司)、888 Titrino自动电位滴定仪(瑞士万通公司)、848 Titrino plus自动电位滴定仪(瑞士万通公司)、ZDJ-3D全自动电位滴定仪 (北京先驱星科技发展有限公司)测定供试品含量,结果含量分别为100.04%、100.53%、99.68%、99.62%,RSD为0.5%;分别使用两厂家共四批次的丙酮 (分析纯和色谱纯各两批)作为溶剂,按2.2.2项下的方法测定,结果分别为 100.26%、99.57%、99.83%、100.37%,RSD为0.4%;分别使用4份不同人员调制的中性丙酮作为溶剂,按2.2.2项下的方法测定,结果分别为100.07%、99.43%、99.78%、100.28%,RSD 为 0.4%。
2.3 两种方法测定结果比较[7]取两批尼美舒利原料药(批号1#,2#),用高效液相色谱法和电位滴定法分别测定含量,结果如表4所示。将两组结果进行F检验,得到在95%置信水平上两批样品F值均小于F临界值,说明两种方法在精密度上无明显差异。进而用t检验分析,结果两批样品t值分别为2.141 和 1.460,均小于临界值 (t0.05/2,5=2.571),表明两种方法测得的尼美舒利含量无统计学差异 (P>0.05)。
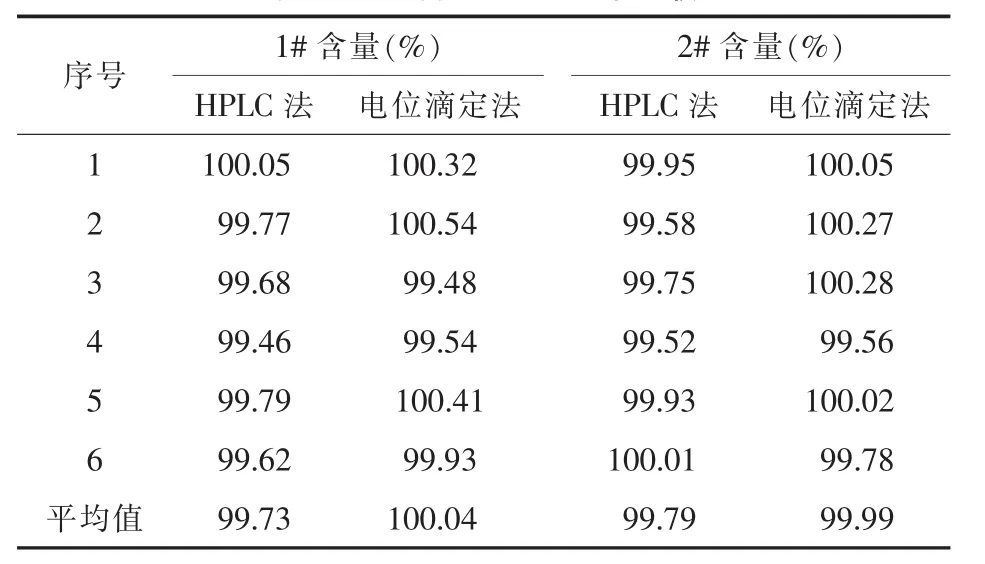
表4 两种方法测定结果比较
3 讨论
3.1 高效液相色谱法 虽然中国、英国等多国药典常采用滴定法测原料药的含量,ICH(international councilpor harmonization,人用药品注册技术要求国际协调会)也认可容量法[8,9],但与容量法相比,HPLC法具有专属性强、灵敏度高、准确性好、取样量少和方便大批量分析等优点。特别是当原料药中存在杂质或储存期降解产生杂质,对滴定造成潜在干扰时,HPLC法更显示出独特的优势。通过对两批尼美舒利原料进行测定,发现HPLC法和电位滴定法测定结果无统计学差异,但HPLC法专属性更强、重复性及耐用性更佳。
3.2 电位滴定法 电位滴定法是容量法中的一种,具有操作简便、分析时间短、准确度高等优点[10]。2017年中国食品药品检定研究院发起了《尼美舒利含量测定能力验证》,检验方法是电位滴定法,参与此次能力验证的实验室为141家,取得满意结果的为 109家[11],通过率 77.30%,低于同年其他项目通过率的平均值,提示该方法可能耐用性较差。该文研究发现,用电位滴定法测定尼美舒利含量时应注意两点:一是滴定参数的设置。空白滴定应设置成MET等量滴定模式,将滴定液增量设置成最小,以防过量,同时应适当调高等当点后体积,以防止得到的是伪终点。二是中性丙酮的配制。方法中使用到中性丙酮 (对酚酞指示液显中性),该次研究发现,当滴定参数设置合理时,丙酮是否调中性对测定结果没有影响,但由于调中性时丙酮变色不明显,容易调过头导致结果偏低。
两种方法均可用于尼美舒利原料药的质量控制,各有优势,采用电位滴定法时应注意参数的设置以及中性丙酮的配制。
