基质固相分散-液相色谱-串联质谱联用技术检测鸡蛋中14种氟喹诺酮类药物残留
2018-11-07郑良清尚吟竹赵晓亚车志龑
罗 静, 郑良清, 尚吟竹, 赵晓亚, 叶 诚, 王 晗, 车志龑, 王 鹏*
(武汉海关,湖北武汉 430050)
氟喹诺酮类药物(Fluoroquinolones,FQs)是一类人工合成的广谱、高效、低毒抗菌药,在畜牧养殖中被广泛用于预防和治疗动物疾病,但其在禽类养殖过程中的滥用,会导致食品样本中累积量急剧递增,对人体健康造成不可预测的潜在风险[1 - 3]。因此,在我国国家标准(GB/T 2007)中对其进行了限量:鸡蛋中西诺沙星、恩诺沙星、洛美沙星、氧氟沙星检出量不得超过0.5 μg/kg;沙拉沙星、诺氟沙星、培氟沙星检出量不得超过1.0 μg/kg;环丙沙星检出量不得超过1.2 μg/kg;依诺沙星检出量不得超过1.5 μg/kg。
目前,食品中喹诺酮类药物残留的检测方法主要有酶联免疫分析法[4]、毛细管电泳法[5 - 6]、高效液相色谱法[7 - 8]、液相色谱-串联质谱法(LC-MS/MS)[9 - 11]和气相色谱-质谱法等。其中,基于LC-MS/MS的检测方法灵敏度高,能对多种喹诺酮类药物进行同时筛选和确证,是氟喹诺酮类药物多组分残留确证检测的最佳方法之一。然而,食品样品中氟喹诺酮类药物的检测一直缺乏合适的样品前处理技术。以鸡蛋样品为例,由于样品基质复杂,氟喹诺酮类药物残留量通常较低,因此目前通常采用溶剂提取的方式进行预处理,步骤繁琐、溶剂消耗量大、且耗时较长。相比之下,基质固相分散(Matrix Solid Phase Dispersion,MSPD)技术[12 - 14]这种适用于固体、半固体样品的前处理方法,具有操作简单快捷、抗基体干扰能力强的特点,非常适合作为鸡蛋样品的前处理手段。
本工作以14种氟喹诺酮类药物为研究对象,建立了一种MSPD-LC-MS/MS分析方法,在简化样品前处理过程的基础上,实现了鸡蛋中14种氟喹诺酮类药物残留的高灵敏检测。
1 实验部分
1.1 仪器、药品与试剂
8050型液相色谱-串联质谱仪(日本,岛津公司);2-16PK型低温离心机(德国,Sigma);MS2型漩涡混合仪(德国,IKA);TurboVap LV型氮吹仪(瑞典,Biotage);ECLIPSE 80i显微镜(日本,Nikon)。
双氟沙星(纯度90.0%)、依诺沙星(纯度99.0%)、氧氟沙星(纯度99.0%)、诺氟沙星(纯度99.1%)、环丙沙星(纯度94.0%)、洛美沙星(纯度98.7%)、达氟沙星(纯度94.0%)、恩诺沙星(纯度99.0%)、沙拉沙星(纯度97.0%)、西诺沙星(纯度99.5%)、氟罗沙星(纯度99.6%)、奥比沙星(纯度97.7%)、司帕沙星(纯度98.8%)、马波沙星(纯度99.0%),均购于Dr.E公司。标准贮备溶液:准确称取10.00 mg标准品,用甲醇溶解并定容至10 mL棕色容量瓶中,若不能完全溶解,加入200 μL甲酸。此溶液浓度为1 mg/mL,4 ℃ 下避光保存,有效期为6个月。将各标准储备液稀释,配成混合标准溶液,各组分浓度均为10 μg/mL。精确吸取适量混合标准工作液用初始流动相逐级稀释,加入到与试样基质相应的阴性样品中,使各目标分析物浓度为0.1~50 μg/kg,所有样品均设置5个平行组。乙酸、乙腈为色谱纯试剂。
1.2 样品前处理
鸡蛋去壳,蛋清和蛋黄搅拌均匀。取1.0 g均质样品置于研钵中,加入3.0 g硅胶,研磨均匀后装入容量为30 mL的注射器内,注射器底部及硅胶上部各垫一张滤纸,压实样品。用30 mL氨甲醇溶液进行洗脱,收集洗脱液,8 000 r/min离心后,取上层清液于45 ℃条件下氮气吹至近干。然后加入1 mL 0.1%乙酸溶液,漩涡混合1 min,12 000 r/min超速离心10 min,取上清液用0.45 μm滤膜过滤,滤液待测。
1.3 色谱条件
色谱柱:Shim-pack GISS C18柱(50×2.1 mm,1.9 μm),流速:0.25 mL/min,柱温:30 ℃;进样量:10 μL。流动相A:0.1%乙酸溶液;流动相B:乙腈。洗脱梯度如下: 0~6 min,95%A;6~8 min,95%~88%A;8~12 min,88%A;12~16 min,88%~75%A;16~18 min,75%A;18~20 min,75%~10%A;20~23 min,10%A;23 min10%~95% A;23~27 min,95%A。
1.4 质谱条件
离子化模式:ESI+;喷雾电压(IS):5 500 V;雾化器(N2):12 mL/min;气帘气(N2):9 mL/min;辅助气(N2)流速:8 mL/min;离子源温度:480 ℃;检测方式:多反应监测(MRM)模式;其他质谱参数见表1。
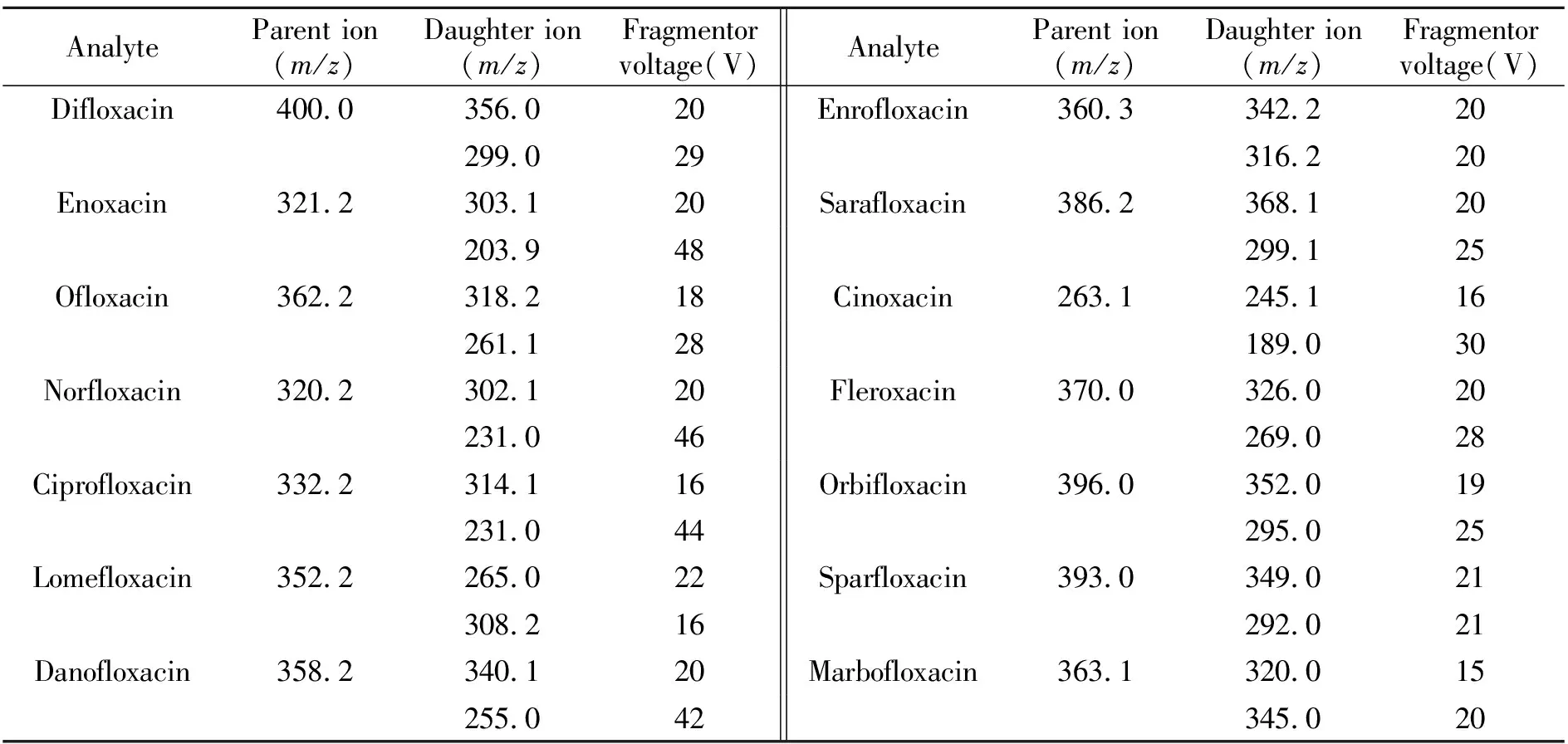
表1 14种氟喹诺酮类抗生素最优化的质谱条件
2 结果与讨论
2.1 样品前处理方法优化
鸡蛋样品中含有大量蛋白质和脂肪,因此样品易乳化,基质干扰严重,样品预处理过程中重现性通常较差,导致喹诺酮类药物残留分析困难。国家标准(GB/T 20366-2006)方法中,采用甲酸-乙腈混合溶液配合均质器均质提取两次,提取液用乙腈饱和的正己烷除脂净化的方式进行预处理;国家标准(GB/T 21312-2007)方法中,采用磷酸盐水溶液超声辅助提取,HLB固相萃取柱纯化的方式进行预处理。虽然以上预处理方法能得到较好的纯化效果,但需要均质器或超声辅助,且耗时长、操作复杂、试剂消耗量大、成本高。为了在保证良好的重现性及较高的提取效率的前提下简化预处理过程,本实验采用了基质固相分散的方法,将适量鸡蛋直接与硅胶混合研磨,使鸡蛋均匀分散于硅胶颗粒的表面,装柱后进行洗脱。
2.1.1洗脱液的选择比较了不同洗脱液对目标物的洗脱效果,按照预处理方法装柱后,分别加入相同体积的洗脱液1(乙腈/乙酸=99/1)、洗脱液2(乙腈/氨水=20/1)、洗脱液3(氨甲醇溶液)进行洗脱,结果表明,氨甲醇溶液的洗脱效果最佳。
2.1.2洗脱液体积的优化为保证14种氟喹诺酮类药物能被完全洗脱,以加标5 μg/kg的空白鸡蛋样品为目标模型,依次用10 mL氨甲醇溶液洗脱5次,采用LC-MS/MS分别对5次洗脱液进行分析。结果表明,洗脱3次后洗脱液中不再有待测物检出,因此选择洗脱液体积为30 mL。
2.1.3基质固相分散提取过程在本工作的基质固相分散提取过程中,固定相硅胶主要起到分散样品的作用。通常,在有机溶剂提取过程中,鸡蛋样品会产生蛋白质凝结(如图1b),导致目标分析物被蛋白包裹而无法被完全提取,因此在国家标准中需采用均质器辅助提取。而根据显微镜成像分析(如图1a),固定相硅胶的辅助使得鸡蛋样品在加入有机溶剂的情况下,依然能够均匀地附着在硅胶颗粒表面,即保证了样品与洗脱溶剂在洗脱过程中依然能够充分接触,从而达到更好的提取效果和提取重现性。

图1 MSPD(a)与液-液萃取(b)后样品的显微成像图Fig.1 Microscope images of egg samples subjected to MSPD(a) and liquid-liquid extraction(b)
图2 空白鸡蛋试样中添加氟喹诺酮类药物色谱图Fig.2 Chromatogram of fluoroquinolones in spiked egg samples(1) difloxacin,(2) enoxacin,(3) ofloxacin,(4) norfloxacin,(5) ciprofloxacin,(6) lomefloxacin,(7) danofloxacin,(8) enrofloxacin,(9) sarafloxacin,(10) cinoxacin,(11) fleroxacin,(12) orbifloxacin,(13) sparfloxacin,(14) marbofloxacin(5 μg/kg).
2.2 液相条件的优化
2.2.1色谱柱的选择由于喹诺酮类化合物的叔胺基和羧基官能团能在水中发生解离,色谱柱固定相表面的残存硅醇基和金属离子可通过氢键或离子交换作用保留喹诺酮类化合物,导致目标分析物色谱峰拖尾、保留时间不稳定或过长,甚至被完全保留在色谱柱上。因此需要选用以高纯硅胶为基体并经端基封闭处理的C18柱作为分离柱。本工作采用Shim-pack GISS C18柱进行了分离实验,可满足色谱分离要求。
2.2.2流动相的选择与优化流动相的pH值对喹诺酮类化合物的分离和保留具有显著影响:(1)喹诺酮类化合物为酸碱两性化合物,其解离状态和在流动相中的溶解性随流动相的pH值而变化;(2)硅胶基键合固定相表面的残余硅醇基的解离程度与流动相的pH值有关,pH>3即可完全解离。为维持流动相的低pH值以抑制硅醇基的解离和保证喹诺酮类化合物在流动相中的稳定溶解状态,达到较好的分离效果,实验中试验了各种流动相。结果表明,0.1%乙酸溶液与乙腈进行梯度洗脱时,能实现各待测物质完好分离。色谱分离情况如图2所示。
2.3 质谱条件的优化
采用1 mg/L的目标分析物标准溶液在正离子模式下进行母离子全扫描,确定各种抗生素的分子离子,然后分别以各种喹诺酮类化合物的分子离子为母离子,对其子离子进行全扫描。喹诺酮化合物的二级质谱中主要的碎片离子峰是喹诺酮药物分子的脱水峰([M+H-H2O]+)、脱羧峰([M+H-CO2]+)以及脱羧后哌嗪环断裂发生结构重排失去CHR的产物离子([M+H-CO2-CHR]+),以沙拉沙星为例,当碰撞电压较小时,主要是脱水峰、脱羧峰,产生m/z368.1和m/z342.2的二级碎片。升高碰撞电压,哌嗪环断裂,产生m/z299.1 的二级碎片,其可能的裂解途径见图3。
图3 沙拉沙星碎片离子可能裂解途径Fig.3 Possible fragmentation pathway of sarafloxacin
选取丰度较强、干扰较小的两对子离子为定性离子。最后以多反应监测(MRM)正离子模式优化各种质谱参数,最优参数列入表1。
2.4 分析性能
电喷雾电离离子源(ESI)易受样品基质的影响,实验中发现鸡蛋样品的基质会对喹诺酮类药物的离子峰信号产生增强效应,因此我们采用向阴性鸡蛋样品中添加标准品的方式进行定量。结果表明,14种氟喹诺酮药物的检出限为0.1 μg/kg,方法线性范围为0.1~50.0 μg/kg,相对标准偏差(RSD)在8.8%~10.4%之间(c=0.5 μg/kg,n=3)。
2.5 回收率与精密度
在与检测样品基质相同的阴性样品中添加不同浓度的氟喹诺酮标准品,每个水平做5个平行样,测定结果见表4。空白样品中添加最低浓度的14种氟喹诺酮类药物的标准溶液,以3倍信噪比为检出限,计算得出14种氟喹诺酮类药物检出限达到0.1 μg/kg。

表2 平均回收率及精密度(n=5)
2.6 实际样品的测定
采用本工作建立的方法对湖北出入境检验检疫局接检的50个样品进行分析,结果有两个鸡蛋样品中恩诺沙星的残留量超出限量标准,残留量分别为1.10 μg/kg和4.95 μg/kg。由检测结果可知,阳性样品检出率较低,证实了市场上大部分鸡蛋中的喹诺酮类药物残留未超出限量标准。
3 结论
本文建立了一种基质固相分散-液相色谱-质谱联用检测14种残留氟喹诺酮类药物的新方法,具有快速、简便和高灵敏的优点,适合于固体及半固体食品样品,特别是鸡蛋样品中多种氟喹诺酮类药物的高通量分析,相较于国家标准方法优势明显。
