儿童扩张型心肌病的临床特点及1例家族性病儿的基因突变分析
2018-09-26
(广西医科大学第一附属医院儿科,广西 南宁 530021)
扩张型心肌病(DCM)是儿童和青少年心源性猝死的常见病因[1]。儿童心肌病约占儿童心脏病的1%[2],DCM是儿童心肌病中最常见的类型,病因复杂,包括心肌炎、神经肌肉疾病、家族性心肌病、遗传代谢病等[3],预后差,仅有30%~40%病儿可明确病因[4-5]。本研究分析了49例DCM病儿的临床特点,并分析了1例家族性DCM的基因突变结果。现将结果报告如下。
1 资料与方法
1.1 一般资料
收集2006年1月—2016年1月于我院儿科首次住院治疗的DCM病儿49例,男22例,女27例;起病年龄6个月~14岁,平均起病年龄为(89.2±49.2)个月;其中<1岁者4例,1~5岁者13例,>5岁者32例。
1.2 研究方法
所有DCM病儿均详细询问病史,体格检查,收集病儿的年龄、性别、临床症状、体征、肌酸激酶同工酶(CK-MB)、心肌钙蛋白I(CTnI)、心电图、胸片、超声心动图以及基因检测结果,部分病儿行心脏CT或者心脏核磁共振检查。入选标准:诊断符合1995年WHO/ISFC心肌病分类标准和中国心肌病诊断与治疗建议工作组制定的心肌病诊断建议中的标准[6-7];年长儿心功能评估采用NYHA心功能分级法,婴幼儿采用改良的Ross评分法。采集家族性DCM先证者、先证者父母、先证者妹妹、先证者哥哥的静脉血,采用一代测序法(Sanger法)完成基因测序及变异位点的筛查,确证变异位点的参考数据库为HGMD Pro、PubMed、1000Genomes和dbSNP。基因检测和数据判读由北京康旭医学检验所完成。49例病儿出院后门诊随访或电话随访,随访时间至2016年12月,以死亡或心脏移植作为随访终点。
1.3 统计学方法

2 结 果
2.1 DCM病儿的临床基线资料
49例DCM病儿入院时主要的症状和体征为咳嗽、气促、乏力、肝脏增大以及心音低钝等,其中40例(81.6%)病儿心胸比增大,大部分病儿合并呼吸道感染,但无栓塞事件发生。见表1。
2.2 DCM病儿的心脏超声检查结果
49例DCM病儿左心室均增大,左心室舒张末期内径(LVDd)平均值为(57.4±8.9)mm,左心室收缩末期内径(LVDs)平均值为(47.6±8.3)mm,左心室射血分数(LVEF)<30%者16例(32.7%),左心室短轴缩短率(LVFS)<20%者31例(63.3%)。所有病儿均存在不同程度的瓣膜反流,但仅1例为重度瓣膜反流;25例(51.0%)病儿存在肺动脉高压,其中仅1例为重度肺动脉高压;20例(40.8%)病儿存在少量心包积液,3例(6.1%)病儿出现左心室附壁血栓。
2.3 DCM病儿的随访情况
病儿均给予强心剂、利尿剂和ACEI等治疗,合并感染者给予抗感染治疗。首次住院病儿无死亡者,出院后获得随访32例,随访时间1~131个月,平均29.25个月,死亡13例,5例病儿出院后坚持规律服用强心和利尿药半年~1年后停药,但至随访时生活质量可,无明显运动受限;14例病儿目前坚持规律用药,用药时间2~5年,日常活动无明显影响;余17例失访。13例死亡病儿均因心力衰竭进行性加重而死亡,无猝死者,无心脏移植者。Kaplan-Meier生存曲线显示,病儿1、3和5年生存率分别为68.2%、54.4%和54.4%,诊断后2年内死亡风险最高。见图1。
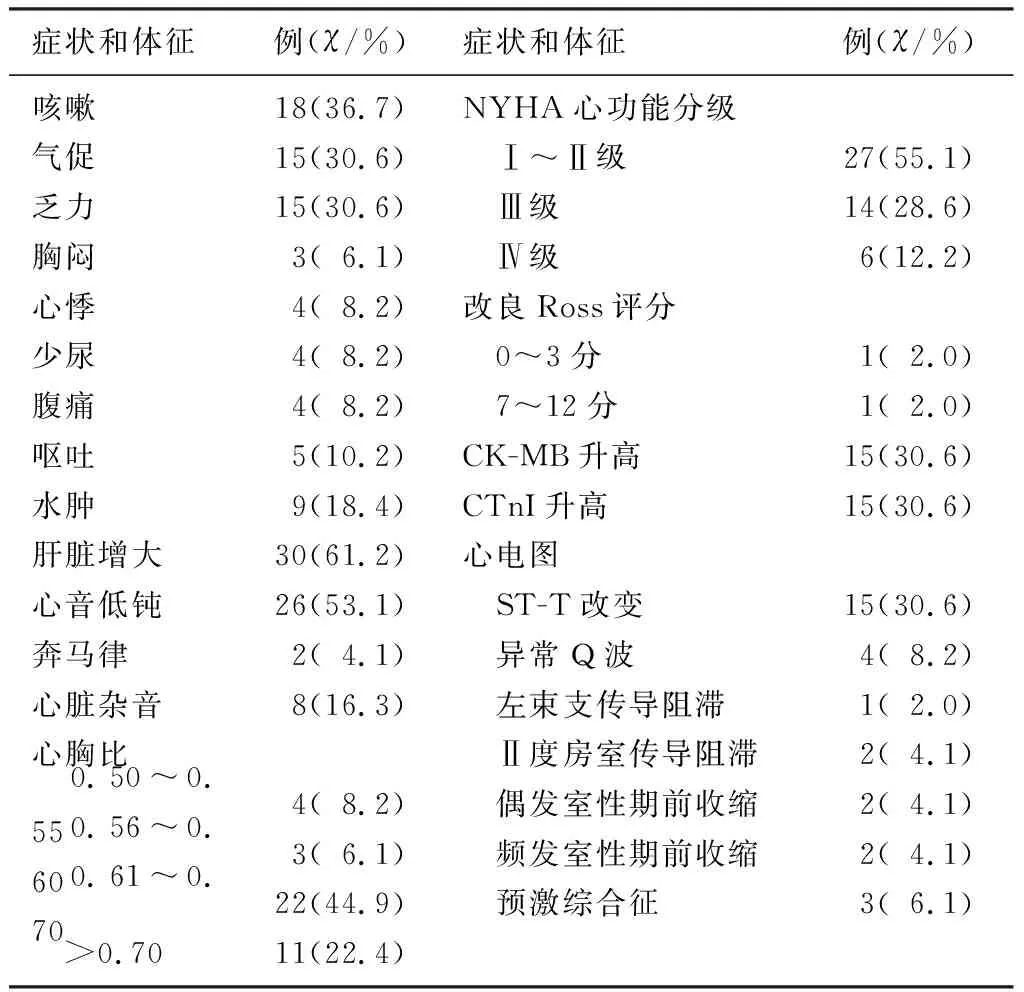
表1 DCM病儿的临床基线资料

图1 DCM病儿的Kaplan-Meier生存曲线
2.4 1例家族性DCM基因检测结果
先证者,男,12岁,表现为呕吐、乏力、尿少,体检心律不齐、心音低钝,肝脏肋下触及2 cm。X线胸片示心胸比为0.7,肺炎改变。24 h动态心电图示:窦性心律不齐;频发室性期前收缩,共7 226次,多源,部分成对,呈联律、间位,占总心搏的5.4%;交界性期前收缩,不完全性干扰性房室脱节;间歇性ST段改变;心率变异性中度降低。胸部B超检查示双侧胸腔积液。心脏彩超检查示:左房室明显增大并室壁整体运动弥漫性减弱,左心室近心尖部丰富肌小梁回声,二尖瓣与三尖瓣轻度反流;LVDd 64 mm,LVDs 57 mm,IVSd 7 mm,LVPwd 7 mm,LVEF 23%,LVFS 11%。发病后10个月因心力衰竭死亡;其姐于10岁时在外院诊断为DCM,诊断后数月因心力衰竭死亡。其三代家系图谱见图2。
先证者的家庭成员的基因突变位点见表2,先证者的家庭成员均无临床症状及体征。先证者的DMD 基因出现 c.2473T>G的核苷酸变异,该变异导致了第825号的氨基酸由Trp变为Gly(p.Trp-825Gly);先证者TTN基因出现c.32186C>G的杂合核苷酸变异,该变异导致了第10729号的氨基酸由Thr变为Arg(p.Thr10729Arg);先证者SCN5A基因出现了c.2663T>C的杂合核苷酸变异,该变异导致了第888号的氨基酸由Phe变为Ser(p.Phe888Ser);先证者RBM20基因出现c.3512C>T的杂合核苷酸变异,该变异导致了第1171号氨基酸由Thr变为Met(p.Thr1171Met)。见图3。先证者上述基因的突变均为错义突变,非多态性变化。

:男性先证者;:女性死亡者。
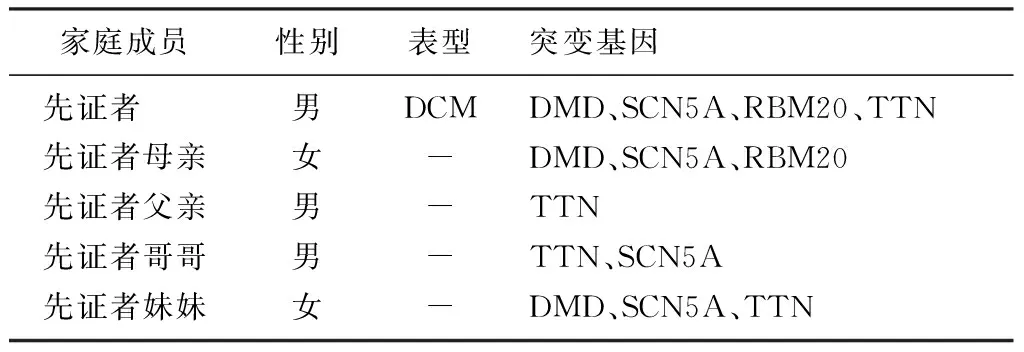
家庭成员性别表型突变基因先证者 男DCMDMD、SCN5A、RBM20、TTN先证者母亲女-DMD、SCN5A、RBM20先证者父亲男-TTN先证者哥哥男-TTN、SCN5A先证者妹妹女-DMD、SCN5A、TTN
3 讨 论
DCM病儿发病年龄各家报道不一。美国多中心调查显示,≤1岁婴儿DCM的发病率明显高于1岁以上的儿童[8],国内韩燕燕等[9]对62例DCM病儿的临床资料进行分析发现,诊断年龄4.0~6.9岁者15例,7~14岁者34例,年长儿占79%;但是章旭等[10]的研究结果显示,≤1岁的DCM病儿发病率最高,段庆宁等[11]的研究亦显示DCM以≤2岁的儿童发病为主。分析发病年龄不一的原因可能与各研究的纳入标准、样本量及地域等因素有关。本组中DCM病儿<1岁者4例,1~5岁者13例,>5岁者32例(65.3%),提示广西地区DCM的发病可能以年长儿居多。LIPSHULTZ等[8]的美国多中心儿童心肌病研究结果显示,DCM的男女比例为1.5∶1,年发病率男孩高于女孩;NUGENT等[12]调查结果显示,澳大利亚儿童心肌病男女比例为1.12∶1。国内也有小样本资料显示DCM病儿男性多于女性。本组DCM病儿中女性多于男性,与文献报道结果不一致,推测其原因可能与样本量小、广西地区差异、性激素影响及X染色体突变等有关。

图3先证者的基因突变
DCM病儿就诊时多表现为心功能不全,少部分病儿出现严重心力衰竭;常合并呼吸道感染,甚至肺炎;部分病儿就诊时已有CK-MB或者CTnI升高。DCM病儿心电图以ST-T段改变、异常Q波、室性期前收缩等异常为主,超声心动图常提示心腔扩大,收缩功能减弱,少部分病儿合并心腔内附壁血栓等。DCM的临床诊断并不困难,超声心动图能直接观察心脏形态学和血流动力学的变化,具有准确、便捷、无创、重复性好、可动态观察、无电离辐射等优点,是诊断DCM的首选方法[13]。
DCM预后的相关危险因素目前尚无统一标准,有研究表明发病年龄>5岁是儿童DCM的独立危险因素[11],但ALVAREZ等[14]则认为发病年龄>6岁、合并先天性心脏病以及起病时LVEF低是儿童DCM的独立危险因素。目前文献报道的DCM生存率差异较大。国内有研究显示DCM病儿的5年生存率为44.79%~48.00%[10,15];TSIRKA等[16]随访了91例DCM的病儿,其5年的生存率为83%;TOWBIN等[4]对美国和加拿大1 426例DCM病儿进行了系统研究,其5年病死率与心脏移植发生率为46%。本组DCM病儿的1、3和5年生存率分别为68.2%、54.4%和54.4%,其诊断后2年内病死风险高。但由于本组DCM病儿的失访率较高,因此其精确的长期生存率仍需进一步评估。但规范的药物治疗能改善DCM病儿的症状,提高生活质量。
DCM的病因复杂,最常见的是心肌炎[17-19]。心肌炎引起DCM的病理生理机制涉及免疫炎症反应导致的心肌纤维化、心室重构、心力衰竭、心肌局部微环境的改变、胶原合成和分解动态平衡间的互相作用等。除强心、利尿、ACEI类等抗心力衰竭、抗心律失常治疗外,炎症性DCM免疫抑制治疗也具有一定的疗效[20]。YOSHIKAWA等[21]证实了使用新型色氨酸柱的免疫吸附治疗能改善由DCM引起的难治性心力衰竭病人的症状和运动耐受能力,在具有高自身抗体评分的病人中特别有效。炎症性DCM的预后较家族遗传性DCM的预后好[4]。本研究中2例家族遗传性DCM病儿发病后病情进展快,均预后不良。因而明确病因对治疗及判断预后有重要价值。
儿童DCM中超过40%的病儿具有家族遗传倾向[22],一个家系中包括先证者在内有≥2例DCM病人可诊断为家族性DCM[23]。其主要遗传方式为常染色体显性遗传,其次为性连锁遗传、常染色体隐性遗传及线粒体遗传。目前已发现超过40个不同的基因突变可导致DCM[24-26]。杜兴氏肌营养不良基因(DMD)是人类最大的基因,其突变可导致细胞骨架结构的异常[27],主要引起3种疾病,即DUCHENNE型肌营养不良(DMD)、BECKER型肌营养不良(BMD)以及X连锁家族性DCM(XLFDCM)。XLFDCM是一种主要侵犯年轻男性的DCM,常呈快速进展的充血性心力衰竭,一般不出现骨骼肌疾病的临床症状[28-30]。HOOGERWAARD等[31]在研究中发现,DMD以及BMD家系中的女性携带者大约22%出现DCM,其机制可能是X染色体失活;刘剑等[28]在X-连锁DCM病人DMD基因突变分析及临床评价中发现,4例女性携带者中1例为DCM,1例呈早期心肌病的表现,提示DMD基因突变所致XLFDCM家系中的女性携带者也有患病的可能。本组先证者DMD c.2473T>G突变导致第825号氨基酸由Trp变为Gly(p.Trp825Gly)的错义突变,遗传来源于先证者母亲,先证者母亲为杂合子,先证者为半合子,符合X连锁隐性遗传方式;先证者母亲是杂合子,至今无心肌病表现。先证者姐姐10岁时因DCM死亡,虽缺乏其基因测定结果,但我们推测病儿的姐姐可能是杂合子,突变来自于先证者母亲,可能因X染色体失活而发病;先证者妹妹也携带该基因,有DCM的发病风险,建议对其进行长期的随访观察。先证者家系的特点符合XLFDCM,从家系基因检测结果中可以推断,DMD c.2473T>G(p.Trp825Gly)突变可能是导致先证者发病的致病突变,但需进一步行全面的家系基因筛查及对照研究加以验证。
应该值得注意的是,本研究中本例先证者检测到的TTN c.32186C>G(p.Thr10729Arg)、SCN5A c.2663T>C(p.Phe888Ser)以及RBM20 c.3512C>T(p.Thr1171Met)的杂合错义突变,虽均为非多态性变化,但其致病性尚未见文献报道;鉴于绝大多数心肌病是单基因病[32],因而这些基因突变在本家系中的作用有待进一步研究。随着遗传学技术的进展及二代基因测序的出现,为DCM的精准诊断提供了强有力的工具,如何对测序数据进行分析,识别出真正的致病突变成为新的挑战[33-34]。
