木村病四例报道及诊断思路分析
2018-09-03刘娟姜丽丽王友莲
刘娟,姜丽丽,王友莲
木村病(Kimura's disease,KD),也可称为嗜酸粒细胞增生性淋巴肉芽肿、软组织嗜酸性肉芽肿、金氏病等,首先由金显宅等在1937年发表的病例报道中以“嗜酸粒细胞增生性淋巴肉芽肿”出现,1948年日本学者木村(Kimura)等对该病进行了较为详细的描述后被称为KD[1],是一种临床罕见的、病因不明的慢性自身免疫性疾病,容易被漏诊、误诊。本文报道江西省人民医院明确诊断的KD 4例,并复习相关文献,旨在提高对该病的认识,为早期诊治提供参考。
1 病例简介
1.1 一般资料 收集2014年2月—2017年4月在江西省人民医院经组织病理检查确诊的4例KD患者的临床资料,其中,男3例,女1例;发病年龄为15~61岁,平均发病年龄为40.5岁;病程为3~30年,平均病程为12.5年。
1.2 临床表现 4例患者均表现为局部包块,质韧,呈多发性;2例患者伴包块处皮肤瘙痒,1例患者伴全身皮肤瘙痒,1例患者伴包块红肿;4例患者均无疼痛、压痛、压迫症状等不适;1例患者头晕、头痛、口干、耳鸣不适;4例患者初发症状均为头颈部局部肿块,肿块直径以2.0~3.5 cm居多,1例患者伴双侧腹股沟、双侧腋窝肿块,2例患者累及唾液腺(腮腺),1例患者查体可触及局部浅表淋巴结肿大;4例患者既往均体健。
1.3 辅助检查 4例患者血常规示外周血嗜酸粒细胞分数0.20~0.34(参考范围:≤0.05),嗜酸粒细胞绝对值为(2.0~3.1)×109/L(参考范围:≤0.5×109/L);3例患者检测血清免疫球蛋白E(IgE)水平为2 420~10 500 U/ml(参考范围:≤156 U/ml);1例患者血肌酐为116 μmol/L(参考范围:41~73 μmol/L)。1例患者尿常规结果为尿蛋白阳性,24 h尿蛋白定量为1.60 g(参考范围:<0.15 g)。2例患者行骨髓穿刺检查,结果显示嗜酸粒细胞分数分别为0.16、0.12(参考范围:0.03±0.02),2例患者未行该项检查。4例患者影像学表现均显示头颈部或腹股沟淋巴结增大。4例患者的一般资料及实验室检查结果见表1。
1.4 病理 4例患者均行组织切除活组织病理检查,其中局部软组织包块2例,淋巴结2例;病理检查镜下均表现为大量嗜酸粒细胞浸润,伴淋巴细胞、纤维组织增生,并淋巴小结形成。
1.5 治疗及转归 2例患者在外院多次行手术包块切除治疗,1例患者在外院行单纯放疗;3例患者均有复发病史(于治疗后1~5年内复发)。患者1拒绝进一步治疗。患者2入院后予口服泼尼松30 mg、1次/d(随诊减量),口服来氟米特10 mg、1次/d;随诊1年局部肿块逐渐减小,病情稳定,未复发。患者3出院后予口服泼尼松40 mg、1次/d(随诊减量),口服霉酚酸酯50 mg、2次/d,随诊3个月局部肿块明显减小,病情稳定,未复发。患者4于2014-05-22、2014-05-29静脉滴注环磷酰胺800 mg、1次/d,口服泼尼松15 mg、1次/d(随诊减量);于2014-06-16予静脉注射长春新碱2 mg、吡柔比星60 mg化疗,间隔21 d为1周期,共5个周期;于2014-06-30予静脉滴注环磷酰胺800 mg、吡柔比星50 mg化疗1次;于2014-07-30予放疗总剂量36个Gy,分18次给予;住院治疗期间局部肿块明显缩小,嗜酸粒细胞分数下降至参考范围;随访1年,病情平稳。
2 讨论
KD是一种病因不明的、罕见的自身免疫性疾病,有明显的地域性,病例报道主要集中在亚洲地区,好发于中青年男性,男女比例为(3~6)∶1[2]。KD发展缓慢,常需要数年时间,据报道从影像学检查发现小结节到触及肿块的时间达1~10年[3]。本文4例患者的发病人群性别差异、年龄、病程均与文献报道[2-3]相符。
2.1 临床表现 KD以头颈部无痛性、伴或不伴瘙痒、单个或多个皮下软组织肿物为特点,起病隐匿、长期存在、反复发作,常累及头颈部淋巴结及唾液腺体[4],易与颈部的淋巴结结核、肿瘤等混淆;累及眼睑、眼眶、口腔、躯干、四肢等部位的KD也可见报道[5];研究认为肿块的大小与嗜酸粒细胞分数升高程度相关[6]。KD可出现其他脏器受累,合并结缔组织病[7]、肾损害[3]、哮喘[8]、溃疡性结肠炎[9]、血栓闭塞性脉管炎[10-11]、心肌炎[12]等的病例报道均可见,其中以肾损害最为常见[3],LEE等[13]研究指出,10%~60%的KD患者可出现肾损害,其中2/3的患者可表现为肾病综合征,甚至部分患者肾损害可达终末期肾病,需要透析治疗[14]。
2.2 辅助检查 实验室检查结果显示,KD患者外周血嗜酸粒细胞分数及IgE特异性升高,诸多文献报道均可见该特点[15-16];影像学检查如超声、CT、磁共振成像(MRI)可以帮助明确疾病累及范围和进展程度;但有研究认为KD影像学检查结果缺乏特异性[6],影像学表现以不规则局部组织肿块和唾液腺肿大为特点,边界不清晰;MRI可以观察到病变的浸润范围和脂肪组织的弥漫性萎缩[17]。KD需要活组织病理检查最终确诊,但影像学检查对排除恶性病变有一定帮助。病理检查是KD诊断的主要依据,其病理特点为淋巴结正常结构保存、生发中心活跃增生、大量嗜酸粒细胞浸润,多形成嗜酸微脓肿,并可见血管增生样反应[18]。
2.3 发病机制 KD的病因与发病机制不明,但过敏、特异性反应、自身免疫和感染被认为是可能的危险因素[15]。目前多认为KD发病与上述危险因素的触发导致T细胞免疫调节改变,引起以IgE介导的Ⅰ型超敏反应相关[19],同时,辅助性T细胞2的活化可释放多种细胞因子〔如粒细胞巨噬细胞刺激因子(GM-CSF)、白介素(IL)-4、IL-5、IL-13等〕,并促进嗜酸粒细胞的增生和血清IgE的生成。
2.4 诊断过程 4例患者在入院后均进行详细病史询问(主诉、现病史、既往史、过敏史、个人史及家族史,并仔细询问局部肿块首次出现的时间及位置,症状持续时间,是否进行性增大,有无伴随红肿、瘙痒、破溃等及其他系统伴随症状);体格检查包括基本生命体征(体温、脉搏、血压、呼吸)以及全身各组织器官相关检查,重视局部肿块及淋巴结触诊(包括部位、大小、数目、硬度、活动度、粘连、压痛、皮温等)。辅助检查:(1)实验室检查:血、尿、便常规、生化(肝肾功能、电解质、心肌酶谱、空腹血糖、血脂)、免疫系列〔抗核抗体、抗可溶性抗原抗体(ENA)谱、抗瓜氨酸肽抗体、免疫球蛋白、红细胞沉降率、C反应蛋白〕、甲状腺功能、肿瘤标志物;(2)影像学及其他检查:心电图、胸部正侧位X线片、心脏彩超、局部肿块或淋巴结彩超、CT或MRI检查;必要时请相关科室会诊,积极处理。
4例患者完善相关检查后,初步排除感染、肿瘤、结缔组织病、内分泌代谢等疾病;同时发现,4例患者主要临床表现均为头颈部局部缓慢增长的皮下软组织肿块,血常规结果显示4例患者嗜酸粒细胞分数及绝对值均异常升高,3例患者血清IgE水平显著升高,2例患者出现肾脏受累(表现为血肌酐或尿蛋白异常升高);影像学检查显示4例患者均有淋巴结受累,2例患者累及双侧腮腺。4例患者在完善上述无创性检查后诊断仍不明确的情况下,临床医生均选择了取局部肿大组织或淋巴结行活组织病理检查,4例病理检查报告均提及大量增生的淋巴细胞及纤维组织,并淋巴小结形成,周围大量嗜酸粒细胞浸润,提示KD。4例患者的临床表现、实验室检查特征、病理检查结果基本与文献报道[4,15-16,18]的KD一致。
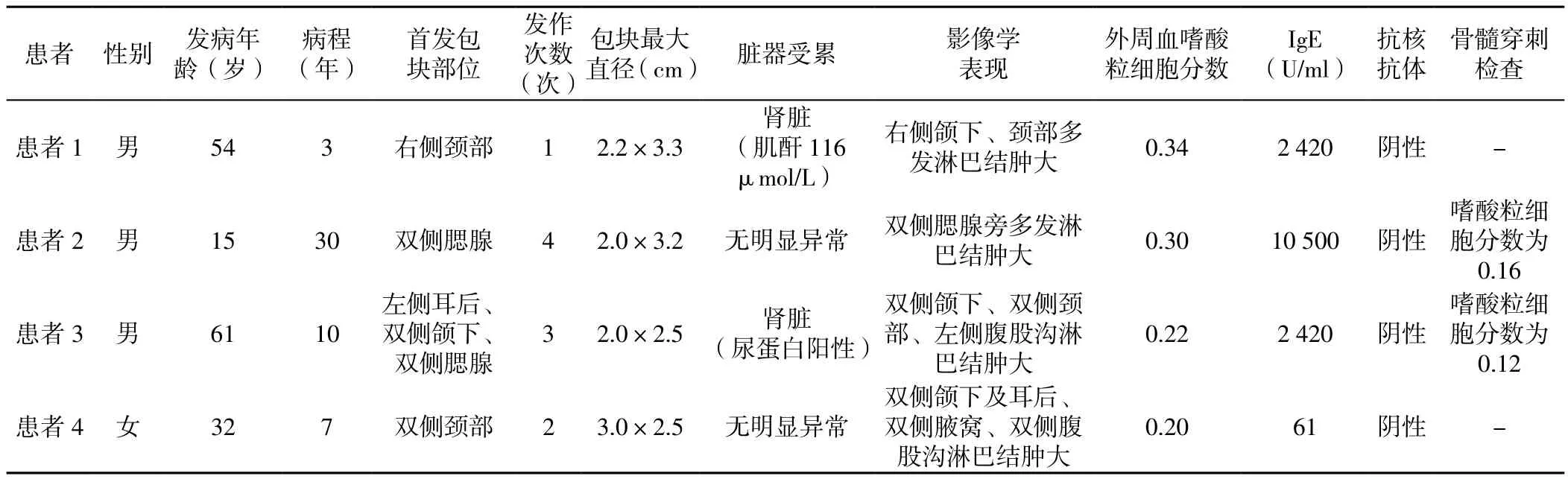
表1 4例患者一般资料及辅助检查结果Table 1 General data and auxiliary examination results of 4 patients
2.5 鉴别诊断 KD较为罕见,目前缺乏统一的诊断标准,对于中青年男性出现头颈部皮下结节伴嗜酸粒细胞、血清IgE升高时,应高度警惕此病,确诊主要依据为活组织病理检查。在鉴别诊断方面,对于淋巴结肿大患者应注意区分病因,包括感染(致病微生物引起的急慢性炎症如急性蜂窝织炎、化脓性扁桃体炎、结核)、肿瘤(淋巴瘤、急慢性白血病、浆细胞肿瘤、转移瘤)、反应性增生(坏死性增生性淋巴结病、血清病及血清病样反应、结缔组织病)、细胞代谢异常(朗格罕组织细胞增生症、脂质沉积病、结节病)等。其中血管淋巴样增生伴嗜酸粒细胞增多症(angiolymphoidhyperp-lasia with eosinophilia,ALHE)与KD有许多相似之处,容易误诊。目前对于KD与ALHE是同一疾病的不同阶段、还是两个独立的疾病仍存在争议。有研究认为两者是不同的疾病,KD代表淋巴组织增生性疾病,ALHE是血管内皮细胞的肿瘤性疾病,伴有继发性炎性反应[15]。两者具有某些重叠的临床和组织学特征,均表现为好发于头颈部的皮下肿块,有复发倾向;实验室检查均可见血嗜酸粒细胞增多,病理学上均有小血管增生,淋巴细胞、嗜酸粒细胞浸润。但ALHE好发于中青年女性,临床表现通常是边界清楚的丘疹,或是红、紫、棕色的结节,可出现结节或皮肤红斑状丘疹,而KD多为深部结节,边界不清,可覆盖皮肤变化;ALHE组织病理表现为血管增生明显,较少出现纤维化,而KD以淋巴小结增生为主,同时伴丰富的嗜酸粒细胞浸润,病变组织通常发生纤维化。此外,嗜酸粒细胞分数、血清IgE水平明显升高、淋巴结肿大、唾液腺受累更多见于KD[16]。文献报道KD可伴随有血清IgG4水平的升高[20],提示应重视KD与IgG4相关的皮肤病变鉴别;1篇关于25例KD患者的回顾性多中心研究提出,KD与IgG4相关性疾病有一些共同的临床病例特征:淋巴结肿大、外周血嗜酸粒细胞增多、血清IgE水平增高[21]。也有研究提出了鉴别点:(1)KD患病人群较年轻,而IgG4相关性疾病多见于中老年男性;(2)IgG4相关性疾病可有全身多个器官和组织的累及,表现为器官肿大;(3)IgG4相关性疾病的实验室检查可见血清IgG4水平显著升高,且在组织病理上可见浆细胞组织浸润,淋巴滤泡形成,并常导致组织增生纤维化和硬化,可见特征性的席纹状纤维化或闭塞性静脉炎[15],上述特点均可与KD相鉴别。
2.6 治疗 目前关于KD的治疗仍无统一的方案,手术切除、单独或联合药物治疗(糖皮质激素、免疫抑制剂)、化疗、局部放疗、IgE抗体治疗均有病例报道[2,22-24]。大多数病例对治疗有良好的初始反应,但复发率高达60%~100%[15];本文3例患者既往经历了局部手术切除或局部放疗后,均出现了复发。一项包括22篇文献、570例KD患者的荟萃分析中,对3种治疗方法(手术切除、放疗、手术切除联合术后放疗)的局部复发率进行了分析,指出手术切除联合术后放疗的局部复发率最低[25]。有报道指出临床上对于小的、单发的、部位易切除的病变,主张手术治疗;对于多发、界限不清或局部浸润以及术后复发的病例主张首选放射治疗[2]。NAKAHARA等[26]报道,皮质激素治疗可以控制KD患者的皮下病变和淋巴结病变,但局部复发常发生在皮质激素逐渐减量的时期,表明仅使用皮质激素治疗并不能治愈KD,因此,在权衡复发和不良反应的风险时,可采用皮质激素治疗作为二线治疗;但SAKAMOTO等[27]认为对于KD最适合的治疗方法应该是口服皮质类固醇,因为该类激素可以减少血清嗜酸粒细胞而不降低血清IgE水平,而高水平的血清IgE对预防面部肿胀和发热起重要作用。在免疫抑制剂方面,环孢素、霉酚酸酯均对该病有效[15]。
2.7 预后 KD属于良性疾病,目前关于该病有恶性增殖趋势的文献报道尚不多见,但其复发率高,复发通常出现在手术、放疗、药物治疗后或者甚至是在肿块减小的药物治疗期间。CHEN等[3]认为该病发病年龄小,高嗜酸粒细胞计数与复发相关,且嗜酸粒细胞计数与肿块的大小及治疗反应相关。有研究提出外周血嗜酸粒细胞计数升高达到白细胞计数的50%以上,血清IgE>10 000 U/ml及唾液腺外出现多个病灶是KD复发的危险因素[28];DENG等[29]对40例术后KD患者进行长期随访,发现治疗后复发KD患者的Ki-67及Notch-1水平较高。有研究认为,IL-21和磷酸化细胞外信号调节激酶1/2表达与疾病的预后相关,其高表达与疾病低复发率相关[30]。KD是一种罕见的、自身免疫相关的慢性炎性疾病,对于临床上头颈部无痛性皮下肿物或者局部淋巴结肿大的患者(尤其是亚洲青年男性),出现周围血嗜酸粒细胞分数或者血清IgE水平升高时,应考虑KD,并积极进行肿大的局部组织或淋巴结活组织病理检查以明确诊断,防止漏诊、误诊;目前未统一该病的治疗首选方案,手术切除、放化疗、激素及免疫抑制剂治疗均是一线治疗方案;KD复发率较高,诊断及治疗后需长期随访。
作者贡献:刘娟、姜丽丽进行文章的构思与设计;姜丽丽、王友莲进行文章的可行性分析,论文的修订,负责文章的质量控制及审校;刘娟进行文献/资料收集与整理,撰写论文,对文章整体负责,监督管理。
本文无利益冲突。
