全基因组混合模型关联分析的极速回归扫描法研究
2018-07-16赵敬丽李淑玲杨润清
赵敬丽,李淑玲,高 进,杨润清,3*
(1.南京农业大学无锡渔业学院,江苏 无锡 214081;2.东北农业大学生命科学学院,哈尔滨 150030;3.中国水产科学研究院生物技术研究中心,北京 100141)
在遗传学领域,全基因组关联分析(Genomewide association study,GWAS)借助单核苷酸多态性(Single nucleotide ploymorphism,SNP)分子遗传标记,在全基因组范围作表型与基因型间相关性分析,发现影响复杂性状基因位点(Quantitative trait nucleotide,QTN)或主效基因。近年来,GWAS在人类复杂疾病性状和动物重要经济性状中应用广泛。受群体分层和个体间亲缘关系等混杂因素影响,表型与基因型间简单回归分析难以适应复杂GWAS。因此,将线性混合模型(Linear mixed model,LMM)引入GWAS[1]。通过剖析由群体分层和个体间亲缘关系所致的混杂偏差,LMM从大量数据中分离真实信号,有效控制QTN定位的Ⅰ型错误率(TypeⅠerror)和Ⅱ型错误率(TypeⅡ error)。使用全基因组标记[2]计算的实现亲缘关系矩阵(Realised relationship matrix,RRM)[1]估计剩余微效多基因方差,基于约束最大似然法(Restricted maximum likelihood,REML)[3]的LMM求解复杂。因此GRAMMAR[4]、 EMMA[5]、 EMMAX[6]、 CMLM[7]、 P3D[7]、FaST-LMM[8]、 GEMMA[9]、 GRAMMAR-Gamma[10]和BOLT-LMM[11]等简化算法相继出现。
大多数改进算法均通过降阶LMM实现简化计算。例如:CMLM法根据实现亲缘关系矩阵将个体聚类成组,将动物模型转化成公畜模型。EMMAX法利用由基因组最佳线性无偏预测(Genomic best linear unbiased prediction,GBLUP)[12]计算的全基因组遗传方差估计剩余微效多基因方差,将线性混合模型转化成当前检验标记效应的剩余误差方差不齐的线性回归模型。GRAMMAR法则将GBLUP估计剩余项作为新表型值并对每个SNP标记作回归分析,此后将剩余微效多基因方差固定为基因组遗传方差,GRAMMAR-Gamma和BOLT-LMM法为对该算法改进。
与REML相比,EMMA法通过对表型值和标记指示变量作谱分解,避免每轮迭代中似然函数计算涉及冗余矩阵运算,成倍量级地提高线性混合模型求解速度。与谱分解每个检验标记EMMA法不同,FaST-LMM法仅需一次谱分解便可检验所有标记。GEMMA算法在谱分解基础上对目标似然函数作二阶求导,寻求全局最优。所有算法均通过预先估计基因组遗传力固定剩余微效多基因方差,提高全基因组高通量标记分析计算效率。虽然与完整(非简化)LMM统计效力相同,但过高估计剩余微效多基因效应,降低表型拟合优度。本研究用剩余多基因遗传力代替方差比值,将多基因遗传力求解限制在(0,1)区间。在FaST-LMM框架内,引入R语言RcppArmadillo程序包[13]中极速线性模型拟合函数(fastLmPure函数)快速估计SNP效应和完整LMM最大似然值。可通过http://th.archive.ubuntu.com/cran/web/packages/RcppArmadillo/RcppArmadillo.pdf查找fastLmPure函数具体用法。
1 理论方法
1.1 基因组混合模型
在校正群体分层等非遗传固定效应后,构建如下含有当前检验SNP加性遗传效应一般线性模型:

y为n个个体校正表型值向量,μ为群体均值,β为被检验标记加性遗传效应,u为随机剩余多基因效应,且u~N(0,),其中K为由遗传标记构建RRM,为未知剩余微效多基因方差,ε为剩余误差效应向量,且 ε~N(0,),其中I为单位矩阵,为误差方差;1为n维列向量,X和Z分别为β和u指示变量矩阵。
在随机效应分布假设下,校正表型值协方差如下:


对K作谱分解即K=USUT,其中S为包含K按降序排列的特征根对角矩阵,U为特征根对应特征向量矩阵。根据UUT=I,协方差矩阵如下形式:
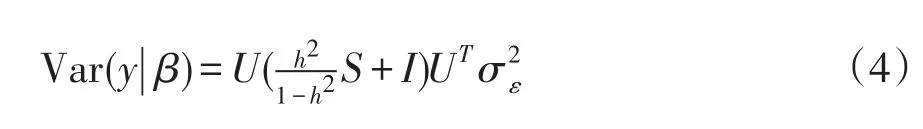
对表型值y和指示变量矩阵X作谱分解转换可得y͂=UTy和 X͂=UT[1 X],则(1)式将变为如下LMM:

因此,通过加权最小二乘法获得β和σ2ε最大似然估计值,表示如下:


对数似然函数进一步简化:

此式关于h2函数。因此,通过对h2在区间(0,1)内一维扫描,优化上述与h2有关对数似然函数,找到h2最大似然估计值。同时,利用与优化遗传力h2相对应β和,统计推断当前检验SNP遗传效应。
1.2 极速回归扫描法
通过搜索剩余多基因遗传力最优估计值,单个SNP的LMM可采用重复加权最小二乘方法实现QTN统计推断。对于高通量SNPs而言,逐个SNP的GWMMAS变成优化每个SNP对应的剩余多基因遗传力全基因组回归扫描问题。为提高GWMMAS计算效率,在求解上述模型(5)时引入极速线性模型拟合函数(fastLmPure函数),加快对式(6)中当前检验SNP效应和LMM极大似然值重复加权最小二乘估计。与常规线性模型拟合函数(lm函数)相比,fastLmPure函数运行速度提高几十倍。fastLmPure函数仅输出当前检验SNP遗传效应和标准差,,2logL,t值和p值等重要统计量需要运行fastLmPure函数后间接计算。
理论上,当前检验SNP剩余多基因遗传力等于性状基因组遗传力和该SNP遗传力(由SNP效应解释的表型方差比例)之间差异。尽管剩余多基因遗传力在高通量SNPs间不同,但仍接近性状基因组遗传力,因为除QTN外的大多数SNPs对数量性状无作用。因此,性状基因组遗传力必须在无效模型(不含SNP效应LMM)下预先估计。在GWMMAS中,从基因组遗传力处出发向下一步或几步扫描,便可快速找到最优剩余多基因遗传力。
每个SNP剩余多基因遗传力被固定为基因组遗传力后,前文中快速回归扫描被简化为EMMAX算法[6],其全基因组扫描速度在不优化剩余多基因遗传力情况下,通过fastLmPure函数达最高值。由于基因组遗传力中已涵盖当前检验SNP变异,因此EMMAX法SNP检测效力略降。该方法估计P值可作为每个SNP快速回归扫描参考。本研究仅选择大效应或较低显著水平(0.05或0.01)SNPs以进一步提高GWMMAS计算效率,优化其剩余多基因遗传力。至此,计算时间复杂度变为O(imn),其中i为全基因组回归扫描迭代次数(1<i≤2)。
1.3 程序设计
在R环境中,按照上述基因组混合模型求解步骤,使用fastLmPure函数执行GWMMAS,执行式(5)线性模型求解。实现亲缘关系矩阵K被计算为K=scale(X)T∙scale(X),其中m为标记个数。求解K特征根S和特征向量,将y和X分别谱变换为y͂和X͂。给定一个剩余多基因遗传力h2,可产生fastLmPure函数因变量y*=和自变量X*=。设计极速回归求解LMM子程序,即fastLmPure函数执行GWMMAS子程序如下:
lmm<-function(ystar,xstar,w){
fit<-fastLmPure(y=ystar,X=xstar)
effects<-fit$coefficients
residual<-ystar-xstar*effects
ve<-var(residual)
std<-fit$stderr
t<-effects[2]/std[2]
p<-2*pnorm(abs(t),lower.tail=FALSE)
logL<-log(det(w))+nobs*log(ve)
}
对每一个SNP,可以从基因组遗传力估计值出发,向下快速搜寻SNP剩余多基因遗传力最大似然估计值。基因组遗传力也适用于EMMAX和GRAMMAR算法。基于无效模型y=1μ+Zu+ε,基因组遗传力快速估计程序由fastLmPure函数编码。如果目标数量性状遗传力来自系谱,基因组遗传力可通过在给定遗传力周围几步扫描快速获得。基因组估计育种值可预测为GV-1(y-1μ),同样适用于GRAMMAR算法剩余项。
2 模拟验证
为评价极速回归扫描法性能,利用玉米基因组数据集模拟验证,数据集可从URL:http://www.panzea.org/#!genotypes/cctl下载。该数据集提供2 648个体的681 258个多态SNPs标记分型结果。从这些标记中随机抽取300 000 SNPs组成标记基因型数据集。将100和1 000 QTNs随机放置在除6和8号以外染色体SNP上。假设被模拟QTNs可解释60%表型变异,从shape=1.66和scale=0.4伽马分布中抽取其遗传效应。同时给定群体均值为0,误差方差为5,再根据模拟QTL基因型随机产生目标性状个体表型值。所有模拟均在一个CentOS 6.5操作系统展开,该系统运行于2.60 GHz的Intel Xeon E5-2660 Opteron(tm)处理器,512 GB内存和20 TB硬盘的服务器。去除数据输入、RRM计算与表型值和标记指示变量谱分解所需时间,极速回归扫描法作回归扫描所需时间为1.986 min,明显低于R语言中lm函数线性模型求解时间(21.289 min)。
2.1 群体分层判别
系谱群体GWAS中群体分层造成目标性状与无关基因间假关联,GWAS假阳性率较高。所谓群体分层(又称为群体结构)是指一个群体中亚群之间在等位基因频率上存在系统性差异。假阳性率是指被错误鉴定为真QTN的假QTN数与实际假QTN数目比率。膨胀系数(基因组控制λGC)可判别群体分层是否广泛适用。λGC是所有SNP检验统计量中位数(或均数)与理论分布中位数比值,实际研究中被定义为所有SNP卡方统计量均值[14]。当λGC≈1时,表明群体分层不存在;当λGC>1时表明存在群体分层或其他混杂变量如家系结构或隐含亲缘关系。Q-Q(Quantile-Quantile)图是将检验统计量可视化的一种标准图形技术,用于判断两个数据集是否来自具有共同分布种群,反映分析模型的统计性质(见图1)。
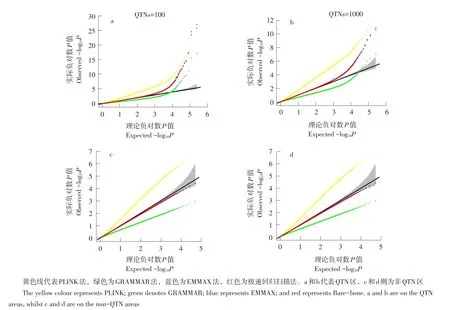
图1 100和1 000 QTN设置下Q-Q图Fig.1 Q-Q plots under the 100 and 1 000 QTN settings
由图1a和b可知,黄色和绿色线严重偏离理论线,表明群体分层存在;对于蓝色和红色线而言,大多数标记观测P值可较好拟合理论阈值线,少数标记严重偏离理论线,表明无群体分层影响,且有QTN存在。
2.2 模型统计性质比较
Liu等介绍3种混杂水平设置下无效分布[15]。本研究采取第3种方法,将遗传标记划分为QTN和非QTN区标记,位于非QTN区标记推导无效分布。在模拟中,将6和8号染色体设置为非QTN区域,该区域包括50 000个SNPs。借助非QTN区域Q-Q图评估是否出现假阴性或假阳性。图1分别展示QTN和非QTN区域Q-Q关系。不论QTN区还是非QTN区,极速回归扫描法和EMMAX法优于PLINK和GRAMMAR法,表明极速回归扫描法和EMMAX法可有效拟合群体分层效应。由非QTN区域Q-Q图可见,PLINK法假阳性率明显偏高,而GRAMMAR法假阴性率较高。这些错误率随模拟QTNs数目增加而升高。
2.3 统计检测效力比较
QTN统计检测效力被定义为检测到QTN数占标记总数比例。图2描述QTN统计检测效力与Ⅰ型错误率关系。PLINK法检测效力与极速回归扫描法相近,但其假阳性率较高;GRAMMAR法检测效力低于极速回归扫描法。尽管EMMAX法与极速回归扫描法Q-Q图几乎重叠,但图2 c和d展示极速回归扫描法检测到比EMMAX法更多QTNs。本研究未与GRAMMAR-Gamma和BOLT-LMM算法比较,因两算法虽可获得与极速回归扫描法相近的统计检测效力,但计算效率较GRAMMAR算法低。

图2 100和1 000 QTNs设置下不同一类错误率统计检测效力Fig.2 Powers against different levels of Type I error under the 100 and 1 000 QTN settings
3 牙鲆生长性状GWMMAS
3.1 GWAS在鱼类经济性状中应用
牙鲆(Paralichthys olivaceus)是我国北方重要海水养殖鱼类,其生长和耐温等多数重要经济性状,均属于数量性状。探索与牙鲆重要经济性状关联基因或QTL,可了解这些性状遗传机制,提高标记辅助育种效率。近年来,GWAS在鱼类等低等脊椎动物经济性状研究中逐渐增多,主要集中于抗病性、抗逆性、发育、性成熟和生长特性等方面[16]。尽管相关研究尚处于初步阶段[17],GWAS已应用于大西洋鲑、虹鳟等少数鱼类生长研究,如Gutierrez等研究大西洋鲑生长相关GWAS,筛选到1个位于Ssa13连锁群SNP位点[18];在虹鳟中,GWAS检测到1个位于omy5位点,可分别解释10、13月龄体重1.4%和1%变异[19]。此外,还有F1代杂交叉尾鮰高温耐受性GWAS,通过EMMAX方法共检测到3个显著性SNP位点[20]。其中,位于14号连锁群位点可解释释表型变异12.1%,另外两个位于16号连锁群SNPs分别可解释表型变异11.3%和11.5%。现有鱼类GWAS研究主要针对单一性状作主效QTN筛选,有关牙鲆鲆经济性状GWAS研究未见报道。牙鲆基因组现已由Shao等测序并组装完成[21],利用基因组信息在全基因组范围内高精度定位牙鲆重要经济性状基因并估计基因组育种值,可为牙鲆经济性状分子遗传解析提供有利条件。
3.2 牙鲆双单倍体群体建立
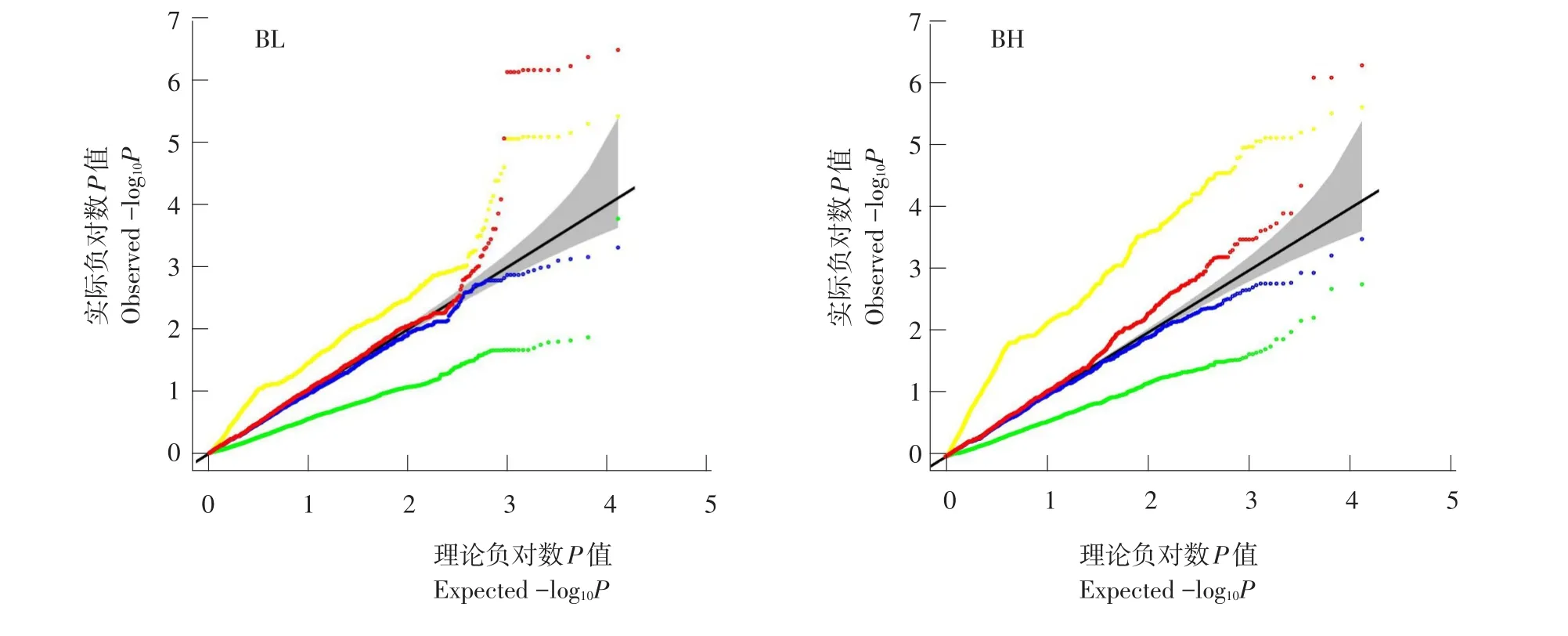
图3 牙鲆2个形态性状Q-QFig.3 Q-Q plots of two morphological traits in Japanese flounder
本研究以威海圣航水产科技有限公司建立的牙鲆育种群作为验证极速回归扫描法性能材料。自2014年开始利用不同来源牙鲆原种群,在两代闭锁繁育基础上,通过静水压方法作雌核发育牙鲆诱导,将紫外线照射灭活牙鲆精子与正常卵受精,受精后静水压处理,诱导染色体加倍,培育双单倍体(DH)牙鲆。按出生时间分为春季和秋季两批,共162个体。4月龄时,对162尾牙鲆注射电子标记,提取鳍条组织DNA。随后将标记后个体混养在6 m×6 m×1 m水泥池,自然光周期下,利用循环水系统养殖,水温5~24℃。养殖期间,每天投喂两次商业化饵料至饱食。自注射电子标记起,电子秤定期称量每个个体体重;在参考标尺下,定期用数码相机从一定高度向下垂直拍摄个体形态性状。直到36月龄,测量2~9次。每次测量前,利用0.05%MS-222将待测个体麻醉,避免处理压力。
根据拍摄图像,利用ImageJ软件获得不同日龄全长、体长、头长、体高和体厚等969个表型记录。采用简化基因组2b-RAD高通量标记分型技术,获得17 297个多态SNP标记,参考牙鲆基因组建立多态标记物理图谱。
3.3 结果分析
分别采用PLINK、EMMAX、GRAMMAR和极速回归扫描4种算法对牙鲆体长(BL)和体高(BH)作GWMMAS,4种算法统计性质比较见图3。与模拟结果类似,PLINK法假阳性率较高,EMMAX法产生一定程度假阴性,GRAMMAR法假阴性程度更高,极速回归扫描法表现最佳。
在GWAS中,通常采用曼哈顿图直观反映所有标记显著性情况。图4~5分别为4种GWAS方法对牙鲆BL和BH基因定位结果。对于BL而言,EMMAX与GRAMMAR法并未检测到任何与该性状相关联基因位点,而PLINK法虽检测到更多高显著水准标记,但并未发现与性状显著关联位点,极速回归扫描法在2和20号染色体上鉴定到可能调控BL基因位点;对于BH,虽然PLINK法与极速回归扫描法均检测到显著位点,但由图5可知,极速回归扫描法检测标记的显著性明显高于PLINK法。该数据充分论证极速回归扫描法在基因定位方面优势,且模型可有效控制QTN定位假阳性率。

图4 牙鲆体长4种比较方法曼哈顿图Fig.4 Manhattan plots of body length in Japanese flounder for the four compared algorithms
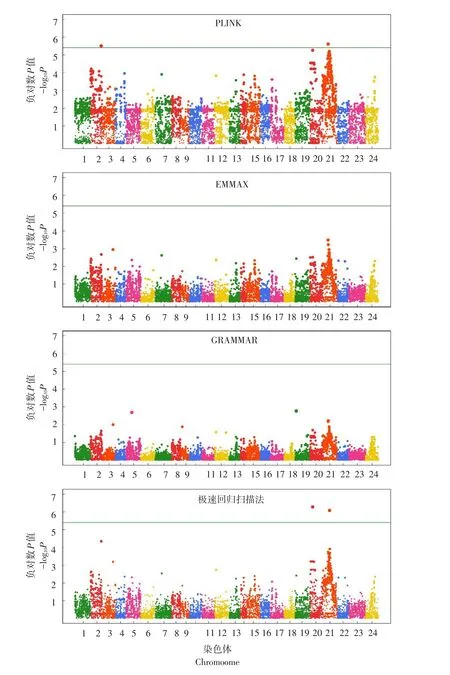
图5 牙鲆体高4种比较方法曼哈顿图Fig.5 Manhattan plots of body height in Japanese flounder for the four compared algorithms
4 结论
在GWMMAS中,无论采用REML法还是谱变换法求解每个标记对应剩余多基因方差,随标记数目增多,计算时间增加。尽管线性混合模型简化求解方法提高计算效率,但均降低剩余多基因方差估计精度及QTN检测效率。本研究提出极速回归扫描法以数量性状基因组遗传力为出发点,探究每个SNP最优剩余多基因遗传力,将GWMMAS转换成高效回归扫描。通过使用R语言Rcp-pArmadillo程序包中极速线性模型拟合函数(fastLmPure函数),缩短计算时间。
