单倍型分析技术研究进展
2018-06-26李双双张迎新范成明陈宇红邓传良胡赞民
李双双,张迎新,范成明,陈宇红,邓传良,胡赞民
1 河南师范大学 生命科学学院,河南 新乡 453007
2 中国科学院遗传与发育生物学研究所,北京 100101
3 中国科学院大学,北京 100049
单倍型 (Haplotype) 是重要的遗传学概念,是指共存于单条染色体上的一系列遗传变异位点的组合。每一条染色体都有自己独特的单倍型,在减数分裂过程中,同源染色体非姐妹染色单体之间的重组可以产生新的单倍型[1]。单倍型包含了一套完整的遗传信息,是描述个体基因组的基础,也是基因组研究必不可少的一个重要方面[2-5]。同源染色体,一个来自母本,一个来自于父本。同源染色体之间不同遗传位点的组合对生物表型有重要影响,如植物中的杂种优势、人类的遗传病等。虽然高通量测序技术、高密度SNP芯片、全基因组测序和基因组外显子测序等全基因组基因型分析技术能在全基因组水平上获得较好的基因组信息,却无法区分同源染色体等位基因的差异,忽略了等位基因间差异对基因表达、功能及其表型的影响[6]。而单倍型可以区分不同亲本染色体的遗传信息,深入了解单条染色体或特定单染色体区域不同遗传位点的组合及遗传,有助于杂种优势的探索及遗传疾病的发病机理的理解和诊断[7]。
单倍型技术主要应用领域包括:在医学上探索致病机理,挖掘致病基因,寻找疾病治疗新方法[8-10];在群体遗传学上分析等位基因间差异[2,11-12],追踪个体亲缘关系[13],了解生物迁徙模式和进化历史[14-16];在农业上发掘优异等位基因变异,探索杂种优势理论等[17-18]。因此,单倍型的研究具有重要的理论意义和实用价值。
本文简要介绍了单倍型分析技术的进展,重点介绍了几个重要技术的原理和应用,对单倍型分析的应用前景、存在的问题和未来发展进行了讨论。
1 单倍型分析技术研究进展
单倍型分析技术主要分为两大类型,间接推断法和直接实验法。间接推断法是借助计算机通过统计学方法,从参考基因组中推断出样本单倍型[6]。随着新一代测序技术的快速发展,人们可以比较容易获得大量的基因组信息,这是间接推断法的基础。直接实验法是指用单分子稀释、染色体微切割和流式分离法等特殊实验方法在一段有限的染色体区域或单染色体获得精确的单倍型信息[3]。表1为重要单倍型分析技术的比较。
1.1 间接推断法
间接推断法根据研究对象的不同又可分为两类:群体推断法[19-20]和家族推断法[21-22]。群体推断法是通过构建一些关联群体的基因池并用统计学方法对预测结果进行分析推断样本的单倍型。如果群体中存在一些突变频率较低的个体,它受连锁不平衡程度的影响往往会被遗漏而无法获得其单倍型信息[23]。家族推断法是根据同一家族众多个体的基因型信息对待测样本进行推断获得其单倍型信息,在使用前要确保同一家族中这些样本基因型信息的可靠性。家族推断法在遗传疾病的研究中有非常重要的作用[24-25]。研究者对一个家庭的父母及其子女四口人进行全基因组测序,经过序列分析可以得知子代基因组精确的重组位点和一些稀有的单核苷酸变异位点。更重要的是,发现该家庭两子女含有米勒综合症和原发性纤毛运动障碍性疾病两个隐性致病基因,对寻找致病基因和疾病治疗方法有重要作用[21]。

表1 重要单倍型分析技术方法比较Table 1 The comparison of mainly haplotype analysis techniques
经文献报道,单倍型推断的方法也多种多样,相继出现了 Clark法[26]、最大期望 (Expectation-Maximization,EM) 算法[33]、相位 (Phase) 法[34]和快速相位 (fastPhase) 法[35]等推断手段[36],其中前3种技术是目前大家普遍使用的推断方法。Clark法是最先产生的单倍型推断技术,根据纯合或杂合的基因型确定已知的单倍型,然后用这些已知的单倍型去和其他杂合待测样本基因型比对,如果该杂合个体的单倍型中有一条和已知的单倍型相同,则相应的另一条单倍型为新的单倍型,循环往复以至找不到新的单倍型为止。最大期望法是把样本各种可能的单倍型都罗列出来并给出一个假定的出现概率,然后通过一步步检测最终确定出待测样本的单倍型。相位法是根据参考样本基因型信息对任意个体通过吉布斯抽样法(Gibbs) 逐步获得杂合样本的单倍型。总体来讲这3种算法中相位算法准确性最好,Clark算法其次,最大期望法居中。然而,它们虽然简便,但根据算法的不同,错误率高达19%-48%[37]。也并不是所有样本的单倍型都能用推断法获得,一些特殊的样本并不适用于这种方法[38]。例如,杂合样本单染色体 SNPs差异的研究和同源染色体之间等位基因的差异分析等[11,28]。
1.2 直接实验法
根据样本自身的特性,直接实验法又可分为两大类:稠密位点单倍型 (Dense) 法[39-42]和稀疏位点单倍型 (Sparse) 法[43-45]。稠密位点单倍型法能精确检测到单染色体局部区域的单倍型,组装结果更完整,在染色体上的排布较密集,是目前最为常用的方法。然而,对于同一染色体上距离较远的单倍型就无法获得。而稀疏位点单倍型法能获得单染色体上几乎所有区域的单倍型信息,但是位点在染色体上的排布比较稀疏,有时不能准确定位该样本单倍型在染色体上的物理位置,甚至会遗漏一些位点[3]。
1.2.1 稠密位点单倍型法
大量文献报道,稠密位点单倍型法能获得染色体上97%的单倍型信息,结果也更精确,应用最普遍。它主要包括单分子稀释法 (Single-molecule dilution)[46-48]、长片段插入克隆法 (Long-insert cloning)[49-50]、保留邻近性转座酶测序法(Contiguity-preserving transposition sequencing,CPT-seq)[51-52]、目标位点扩增 (Targeted locus amplification,TLA)[53]等。这些方法都需要先将样本基因组DNA片段化,再用0.8%琼脂糖凝胶电泳检测高分子量 (High-molecular-weight,HMW) 的DNA片段,最后运用不同的单倍型测序方法获得样本的单倍型信息。以下具体介绍一下单分子稀释法和保留邻近性的转座酶测序法这两种最常用的方法。单分子稀释法是把 HMW DNA随机稀释到 96孔板中组成许多单倍型亚池并对每个单倍型分子用多重置换扩增法 (Multiple displacement amplification,MDA) 扩增,再在所有扩增片段两端加上测序识别标签并进行高通量测序,最后把测序结果按照识别标签序列进行分选并根据参考基因组序列进行拼接组装,得到样本的单倍型序列。它需要借助计算机把小的DNA片段组装成较长的单倍型序列[48]。MDA法是Lizardi等于 2004年创建的一种基于环状滚动扩增的链置换全基因组扩增技术[54]。该技术利用φ29DNA聚合酶的强链置换活性和核酸外切酶活性,以短链寡核苷酸为引物,可对微量 DNA模板进行扩增,并获得浓度达μg级的高质量DNA产物。MDA法不仅扩增效率高于传统的兼并引物扩增法,而且得到的DNA产物片段在kb级,长度也相对均一,能够满足高通量测序的要求。
单分子稀释法由Paul和Apgar教授首次成功运用于人类白细胞抗原位点的研究,对人类疾病的研究具有十分重要的意义,开创了单分子稀释法单倍型测序的先河[28]。2013年,Kaper等用该方法对两名杜氏肌萎缩症患者进行单倍型测序,找到了95%的SNP杂合位点[38]。2014年,Kuleshov教授等用统计学辅助的长序列单倍型测序技术(Statistically aided long read haplotyping,SLRH)对人类全基因组进行单倍型测序,发现了99%的SNP杂合位点,对我们寻找人类基因组上未知的甲基化区域和其甲基化模式有潜在的应用前景,同时对一些差异基因表达机制的研究也有重要作用[55]。然而,该方法也存在一些缺点,如工作量较大,每个亚池稀释到相同的浓度难度很大,具有微弱的扩增偏向性[3]。
保留邻近性的转座酶测序法是利用 Tn5转座酶紧密结合在HMW DNA片段上的特性使短DNA 片段和接头序列紧密结合。首先将带有接头和Tn5转座酶的DNA片段随机分配到96孔板中组成许多单倍型亚池,每个亚池大约含有5%-10%的DNA,然后通过蛋白变性去除Tn5转座酶并通过PCR扩增引入新的标签序列,再将它们混合起来重新分配到96孔板中,这样就随机生成了超过9 200个虚拟隔室,每一个亚池都代表着不同的亚单倍型并进行高通量测序,测序结果按照识别标签序列进行分选并根据参考基因组序列进行拼接组装,得到样本的单倍型序列。该方法的先进性主要体现在以下两个方面:第一,它能通过Tn5转座酶把特异的接头和标签序列同时结合在长链DNA上,使DNA片段不被打乱并保留其邻近序列;第二,它通过转录和PCR两种方法相结合能将很长的单倍型片段分成上千个虚拟隔室,从而使测序结果更精确[29]。此外,该方法能将极短的 DNA片段连接起来并保留其邻近序列,所得样本DNA片段均一性较好,克服了MDA扩增法引起的偏向性。2014年,Amini等首次在Nature Genetics杂志上提出了该方法,他们运用该方法成功获得了人类个体全基因组单倍型序列,由此开发了一种快速稳定高效且操作简便的单倍型测序新技术,为单倍型测序的发展开创了更广阔的空间[29]。随后,Adey教授等在该方法的基础上又结合fragScaff程序把目的序列组装成了更长的片段,N50增加了8–57倍。它能识别并锚定一些新的片段,同时又能剔除一些拼接错误的片段,大大提高了测序的精确度[32]。然而该方法也存在一些弊端,如需要的 DNA量比长片段插入克隆法要多,单倍型组装比较困难。总而言之,保留邻近性的转座酶测序法以其快速、稳定、节约成本的特点深受科学家们的青睐,在未来人类基因组测序和临床医学领域都具有广泛的发展空间。
1.2.2 稀疏位点单倍型法
稀疏位点单倍型法主要包括单染色体测序法(Single chromosome sequencing)[2,56]、单倍型测序法 (HaploSeq)[57]、乳液PCR法 (Emulsion PCR-based methods)[58-59]等。其中单染色体测序是最常见的方法。它是通过单染色体微切割、流式细胞仪分选和微流体分选这3种技术获得样本单染色体,然后用 MDA法进行全基因组扩增再测序,拼接和组装就可得到样本的单倍型信息。它的应用也十分广泛。2010年,Li等用单染色体微切割技术分离人单染色体并进行长片段单倍型测序获得了20 000多个杂合位点的单倍型,其准确性高达98.85%,对检测单染色体SNP变异位点有重要作用[30];同年,Fan等报道了一种新的单染色体分离技术,对人单细胞进行微流体分选获得单染色体,然后进行单染色体单倍型测序获得其单倍型信息,对检测单染色体上SNP差异位点和基因重排有重要作用[2];2011年,Yang等发表了另一种高效的单染色体分离技术,对人用单染色体流式分离技术获得其单染色体,然后用 MDA法进行全基因组扩增再测序,拼接和组装得到了几乎完整的单倍型信息,对人类疾病的研究提供理论依据[56]。总而言之,稀疏位点单倍型法能获得单染色体上几乎全部区域的单倍型信息,是一种较为常用的单倍型获取方法。
1.3 单倍型组装
无论是直接法还是间接法,都涉及到单倍型组装。然而目前常规的单倍型测序技术所得的序列长度较短,不能满足连锁的SNP变异位点样本的单倍型研究,单倍型组装一直是一个难点[60]。图1为个体单倍型组装基本流程[61]。通常个体基因组测序所得的信息是来自于两亲本的,如果对同一SNP位点不同序列分析发现其等位基因信息不同,则它们是来自不同亲本的染色体;反之,它们是来自相同亲本的染色体。就这样依次把所有的SNP位点数据进行分类,拼接组装相互关联起来就构建成了个体单倍型。随后,Bansal等提出了马尔可夫链蒙特卡尔 (Markov chain Monte Carlo,MCMC) 单倍型组装技术。该方法就是把样本的单条或多条序列信息转变为加权图,然后用计算机计算最小和最大切点进行组装,精确性提高20%-25%[61]。同年,Bansal等又推出了一种新的单倍型组装技术:超图分割法 (Hypergraph approximation-Cut,HapCUT)。与马尔可夫链蒙特卡尔方法相比,HapCUT单倍型组装技术算法高效、更精确[62]。尽管该方法可以获得精确组装,但受序列长度的限制,对于一些较长的序列可能无法完全拼接和组装,就不能获得完整的单倍型信息。随后又出现一种基于Hi-C交互信息组装单倍型法,能解决传统方法不能越过着丝粒组装的问题,从而获得更好更完整的单倍型信息[63]。2017年,Edge等在 HapCUT基础上又提出了HapCUT2单倍型组装技术。HapCUT2能对各种不同类型的数据进行组装,与HapCUT相比显著降低了组装的错误率,提高了序列组装的精确性,是目前单倍型组装效率最高的新技术[64]。
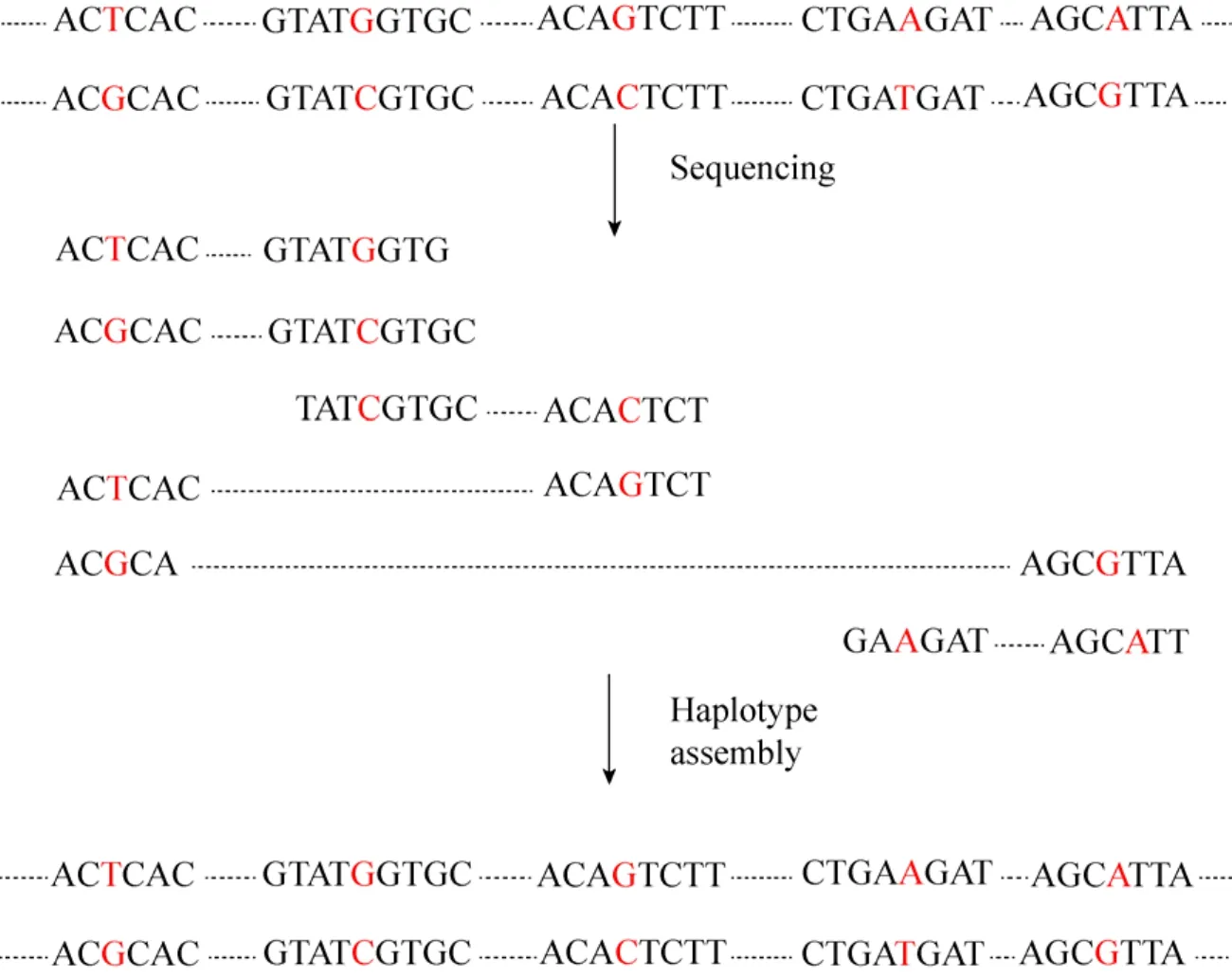
图1 单倍型组装流程图[61]Fig. 1 Flow chart of haplotype assemble[61].
2 展望及应用前景
目前,单倍型分析技术已成为基因组研究领域必不可少的一种重要的研究方法。随着技术的发展,单倍型分析技术也在逐步完善,具有广阔的应用前景。然而每种单倍型分析技术所需的样本类型、物理距离、精确度和实施难易程度等方面都不一样,如间接推断法虽然操作简单,无需进行繁琐的实验,便宜又节省时间,但是准确性较低,错误率有时高达19%-48%;而直接实验法虽然准确性较高,但是过程较繁琐、实验周期较长、费用较高。因此,我们在使用时要根据自己的实验需要对各方面综合考虑,选取最合适的单倍型测序方法。随着长片段测序技术的逐渐成熟以及样品扩增方法的提高,我们预测间接单倍型分析法与直接实验法相结合的技术具有很大的发展空间,既能节约时间又能提高准确性,在未来几年可能被广泛使用[3]。
如今,单倍型分析技术有广泛的应用前景。第一,单倍型技术在人类群体遗传学的研究方面有重要应用。我们通过低精确度的单倍型计算方法就可以获得一些群体特异的单倍型信息,促进人类遗传进化的研究[19]。Schiffels教授对来自不同地区9个人群的基因组进行单倍型测序,最终发现南非人群祖先早在5万年前就与非洲约鲁巴人群发生分离。同时,他们也获得了2 000年前美洲、非洲、东亚和欧洲人类之间的进化关系[65]。Martin等用单倍型技术对来自不同国家人群的潜在致病等位基因进行研究并发现这些致病基因的携带者大部分都是芬兰人,而且它们的基因流动性很大。该研究对单倍型法研究群体进化历史和疾病的研究具有很好参考价值[20]。
第二,单倍型分析技术在植物学领域也有应用,尤其是在水稻[17,66]、玉米[67]和小麦[68]等主要农作物的遗传育种方面有非常重要的作用。杨教授等将高产与普通水稻品种单倍型比较分析发现两者差异较大,高产品种单倍型富含亮氨酸的重复受体激酶基因簇,通过对其结构和功能分析表明该基因簇对优良品种的选育有重要作用。同时,高教授等对不同玉米诱导系品种杂交产生不同基因型的单倍体玉米植株,获得高效诱导玉米单倍体的方法,该方法是目前获得玉米单倍体最为常用的方法[69]。Pozniak教授通过对控制硬粒小麦和普通小麦主干强度的基因进行单倍型分析并精确定位,对小麦育种有重要作用[68]。此外,单倍型分析技术在大豆[18]、苦瓜[70]等植物中也有类似的应用。
第三,单倍型技术还可用于生物理论及医学遗传学领域的研究,探索一些生命现象的本质,从生物学角度解释这些现象并寻找疾病治疗方法。研究者用单倍型技术分析皮肤起痘[71]、脑瘫[72]和耳聋[73]等患者的致病根源,最终发现其都是一种由染色体上杂合变异引起的常染色体隐性遗传疾病[74]。用同样方法,研究者发现由于患者染色体上大量SNPs的交互作用而引起的孟德尔隐性遗传疾病。单倍型技术还可以探索表观遗传与基因调控的相互关系。覃海媚等对鼻咽癌患者和健康人群RTN4基因进行测序和单倍型分析找到了两个致病的核苷酸变异位点,为鼻咽癌的治疗提供理论依据[9]。另外胎儿的单倍型基因组测序检测可用于检测其是否存在潜在的遗传疾病。Kitzman教授等用单倍型测序技术对亲本血浆 DNA深度测序从而获得胎儿单倍型基因组序列,准确率高达99%[75]。
最后,单倍型分析技术有助于完成基因组的精细组装,在基因组学领域有突出作用。例如,对于一些基因组组装不完全的植物或动物,用单倍型分析技术可以准确预测它们的基因组,弥补目前一些领域基因组信息的空缺,对未来基因组学的发展有促进作用[76-77]。随着大规模测序技术的不断发展,单倍型分析技术已取得长足的进步,然而现有技术也存在一些问题,它们在精确度、物理距离以及实现的难易程度等方面并非完全一致,都有局限性。而且仍有很多工作要做,尤其需要探讨的是在重组标记、系谱更大更复杂、标记更多的情况下,如何快速、准确地推断单倍型。更重要的是,目前为止植物领域有关单倍型的研究及应用还很少。单倍型技术对挖掘植物优异等位基因、研究杂种优势理论至关重要,希望未来在这方面能有所突破。
REFERENCES
[1]Kauppi L, Barchi M, Lange J, et al. Numerical constraints and feedback control of double-strand breaks in mouse meiosis. Gene Dev, 2013, 27(8): 873–886.
[2]Fan HC, Wang J, Potanina A, et al. Whole-genome molecular haplotyping of single cells. Nat Biotechnol,2011, 29(1): 51–57.
[3]Snyder MW, Adey A, Kitzman JO, et al.Haplotype-resolved genome sequencing: experimental methods and applications. Nat Rev Genet, 2015, 16(6):344–358.
[4]Jin LN. The correlation analysis method based on haplotype[D]. Changchun: Northeast Normal University,2011 (in Chinese).金丽娜. 基于单倍型的关联分析方法[D]. 长春:东北师范大学, 2011.
[5]Porubský D, Sanders AD, Van NW, et al. Direct chromosome-length haplotyping by single-cell sequencing. Genome Res, 2016, 26(11): 1565–1574.
[6]Lu F. Methods for haplotype reconstruction based on linkage disequilibrium in natural population[D]. Shaanxi:Northwest A & F University, 2005 (in Chinese).鲁非. 利用连锁不平衡信息对推断单倍型算法效能的研究[D]. 陕西:西北农林科技大学, 2005.
[7]Ma L, Li W, Song Q. Chromosome-range whole-genome high-throughput experimental haplotyping by single-chromosome microdissection. Methods Mol Biol,2017: 161–169.
[8]Tewhey R, Bansal V, Torkamani A, et al. The importance of phase information for human genomics. Nat Rev Genet, 2011, 12(3): 215–223.
[9]Qin HM, Wang R, Wei GJ, et al. Association of RTN4 gene polymorphism and haplotype with susceptibility of nasopharyngeal carcinoma in Guangxi Zhuang Population. Cancer Res Prev Treat, 2017, 44(3):221–224 (in Chinese).覃海媚, 王荣, 韦贵将, 等. 广西壮族人群RTN4基因多态性及其单倍型与鼻咽癌易感性的关系. 肿瘤防治研究, 2017, 44(3): 221–224.
[10]Entezam M, Khatami MR, Saddadi F, et al. Genetic analysis of Iranian autosomal dominant polycystic kidney disease: new insight to haplotype analysis. Cell Mol Biol, 2016, 62(2): 15–20.
[11]Hodgkinson A, Grenier JC, Gbeha E, et al. A haplotype-based normalization technique for the analysis and detection of allele specific expression.BMC Bioinformatics, 2016, 17(1): 364.
[12]Loubser S, Paximadis M, Tiemessen CT. Human leukocyte antigen class I (A, B and C) allele and haplotype variation in a South African mixed ancestry population. Hum Immunol, 2017, 78(5–6): 399–400.
[13]Howard NP, Weg EVD, Bedford DS, et al. Elucidation of the ‘Honeycrisp’ pedigree through haplotype analysis with a multi-family integrated SNP linkage map and a large apple (Malus × domestica) pedigree-connected SNP data set. Hortic Res, 2017, 4: 17003.
[14]Wang X, Luo CF, Tao QH, et al. Sequence variation of mtDNA D-loop and origin of labai high-leg chicken.China Poultry, 2016, 38(11): 14–18 (in Chinese).王欣, 罗成峰, 陶清海, 等. 拉伯高脚鸡线粒体 DNA D-loop序列变异与起源分化研究. 中国家禽, 2016,38(11): 14–18.
[15]Tishkoff SA, Dietzsch E, Speed W, et al. Global patterns of linkage disequilibrium at the CD4 locus and modern human origins. Science, 1996, 271(5254): 1380–1387.
[16]Ma XH, Li N, Wang DW, et al. Genetic diversity of the norway rat (rattus norvegicus) in Hainan Island based on mitochondrial cytochrome b gene. Chin J Zool, 2016,51(5): 806–816 (in Chinese).马晓慧, 李宁, 王大伟, 等. 基于线粒体细胞色素b基因分析海南岛褐家鼠种群遗传多样性. 动物学杂志,2016, 51(5): 806–816.
[17]Yi CD, Wang DR, Jiang W, et al. Development of functional markers and identification of haplotypes for rice grain width gene GS5. Chin J Rice Sci, 2016, 30(5):487–492 (in Chinese).裔传灯, 王德荣, 蒋伟, 等. 水稻粒宽基因 GS5的功能标记开发和单倍型鉴定. 中国水稻科学, 2016,30(5): 487–492.
[18]Sheng BH, Liu B, Chen XL, et al. Haplotype analysis and molecular marker development of soybean nematode cyst resistance loci rhg1 and Rhg4 in soybean germplasm.Soyb Sci, 2017, 36(3): 345–350 (in Chinese).盛碧涵, 刘兵, 陈秀兰, 等. 抗SCN位点rhg1与Rhg4在种质资源中的单倍型分析及分子标记开发. 大豆科学, 2017, 36(3): 345–350.
[19]Browning SR, Browning BL. Haplotype phasing:existing methods and new developments. Nat Rev Genet,2011, 12(10): 703–714.
[20]Martin AR, Karczewski KJ, Kerminen S, et al.Haplotype sharing provides insights into fine-scale population history and disease in Finland. bioRxiv, 2017:200113.
[21]Roach JC, Glusman G, Smit AFA, et al. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science, 2010, 328(5978): 636–639.
[22]Elshakankiry NH, Mossallam GI, Madbouly A, et al.Determination of HLA-A, -B and -DRB1 alleles and HLA-A-B haplotype frequencies in Egyptians based on family study. Hum Immunol, 2017, 78: 222.
[23]The genomes project consortium. An integrated map of genetic variation from 1,092 human genomes. Nature,2012, 491(7422): 56–65.
[24]Osoegawa K, Creary L, Mallempati K, et al. Haplotype analyses of classical HLA genes from families. Hum Immunol, 2017, 78: 118.
[25]Catucci I, Casadei S, Ding YC, et al. Haplotype analyses of the c.1027C>T and c.2167_2168delAT recurrent truncating mutations in the breast cancer-predisposing gene PALB2. Breast Cancer Res Treat, 2016, 160(1):121–129.
[26]Clark AG. Inference of haplotypes from PCR-amplified samples of diploid populations. Mol Biol Evol, 1990,7(2): 111–122.
[27]Kruglyak L, Daly MJ, Reeve-Daly MP, et al. Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet, 1996, 58(6): 1347–1363.
[28]Paul P, Apgar J. Single-molecule dilution and multiple displacement amplification for molecular haplotyping.Biotechniques, 2005, 38(4): 553–554.
[29]Amini S, Pushkarev D, Christiansen L, et al.Haplotype-resolved whole-genome sequencing by contiguity-preserving transposition and combinatorial indexing. Nat Genet, 2014, 46(12): 1343–1349.
[30]Li M, Yan X, Hui H, et al. Direct determination of molecular haplotypes by chromosome microdissection.Nat Methods, 2010, 7(4): 299–301.
[31]Adey A, Burton JN, Kitzman JO, et al. The haplotype-resolved genome and epigenome of the aneuploid HeLa cancer cell line. Nature, 2013,500(7461): 207–211.
[32]Adey A, Kitzman JO, Burton JN, et al.In vitro,long-range sequence information forde novogenome assembly via transposase contiguity. Genome Res, 2014,24(12): 2041–2049.
[33]Long JC, Williams RC, Urbanek M. An E-M algorithm and testing strategy for multiple-locus haplotypes. Am J Hum Genet, 1995, 56(3): 799–810.
[34]Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet, 2001, 68(4): 978–989.
[35]Scheet P, Stephens M. A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. Am J Hum Genet, 2006, 78(4): 629–644.
[36]Weale ME. A survey of current software for haplotype phase inference. Hum Genomics, 2004, 1(2): 141–144.
[37]Stephens M, Donnelly P. A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet, 2003, 73(5):1162–1169.
[38]Kaper F, Swamy S, Klotzle B, et al. Whole-genome haplotyping by dilution, amplification, and sequencing.Proc Natl Acad Sci USA, 2013, 110(14): 5552–5557.
[39]Burgtorf C, Kepper P, Hoehe M, et al. Clone-based systematic haplotyping (CSH): a procedure for physical haplotyping of whole genomes. Genome Res, 2003,13(12): 2717–2724.
[40]Dear PH, Cook PR. Happy mapping: a proposal for linkage mapping the human genome. Nucleic Acids Res,1989, 17(17): 6795–6807.
[41]Raymond CK, Subramanian S, Paddock M, et al.Targeted, haplotype-resolved resequencing of long segments of the human genome. Genomics, 2005, 86(6):759–766.
[42]Adey AC. Haplotype resolution at the single-cell level.Proc Natl Acad Sci USA, 2017, 114(47): 12362–12364.
[43]Douglas JA, Boehnke M, Gillanders E, et al.Experimentally-derived haplotypes substantially increase the efficiency of linkage disequilibrium studies.Nat Genet, 2001, 28(4): 361–364.
[44]Yan H, Papadopoulos N, Marra G, et al. Conversion of diploidy to haploidy. Nature, 2000, 403(6771): 723–724.
[45]Vikalo H. Sparse tensor decomposition for haplotype assembly of diploids and polyploids. bioRxiv, 2017:764–765.
[46]Duitama J, Mcewen GK, Huebsch T, et al. Fosmid-based whole genome haplotyping of a HapMap trio child:evaluation of single individual haplotyping techniques.Nucleic Acids Res, 2012, 40(5): 2041–2053.
[47]Kitzman JO, Mackenzie AP, Adey A, et al.Haplotype-resolved genome sequencing of a Gujarati Indian individual. Nat Biotechnol, 2011, 29(1): 59–63.
[48]Bansal V. An accurate algorithm for the detection of DNA fragments from dilution pool sequencing experiments. Bioinformatics, 2017, 155–162.
[49]Steinberg KM, Schneider VA, Graves-Lindsay TA, et al.Single haplotype assembly of the human genome from a hydatidiform mole. Genome Res, 2014, 24(12):2066–2076.
[50]Ciotlos S, Mao Q, Zhang RY, et al. Whole genome sequence analysis of BT-474 using complete Genomics’standard and long fragment read technologies.GigaScience, 2016, 5(1): 8.
[51]Huang M, Tu J, Lu Z. Recent advances in experimental whole genome haplotyping methods. Int J Mol Sci, 2017,18(9): 1944.
[52]Christiansen L, Amini S, Zhang F, et al.Contiguity-preserving transposition sequencing (CPT-Seq)for genome-wide haplotyping, assembly, and single-cell ATAC-Seq. Methods Mol Biol, 2017: 207–221.
[53]Vree PJPD, Wit ED, Yilmaz M, et al. Targeted sequencing by proximity ligation for comprehensive variant detection and local haplotyping. Nat Biotechnol,2014, 32(10): 1019–1025.
[54]Paez JG, Lin M, Beroukhim R, et al. Genome coverage and sequence fidelity of phi29 polymerase-based multiple strand displacement whole genome amplification. Nucleic Acids Res, 2004, 32(9): e71.
[55]Kuleshov V, Xie D, Chen R, et al. Whole-genome haplotyping using long reads and statistical methods.Nat Biotechnol, 2014, 32(3): 261–266.
[56]Yang H, Chen X, Wong WH. Completely phased genome sequencing through chromosome sorting. Proc Natl Acad Sci USA, 2011, 108(1): 12–17.
[57]Selvaraj S, Dixon JR, Bansal V, et al. Whole-genome haplotype reconstruction using proximity-ligation and shotgun sequencing. Nat Biotechnol, 2013, 31(12):1111–1118.
[58]Regan JF, Kamitaki N, Legler T, et al. A rapid molecular approach for chromosomal phasing. PLoS ONE, 2015,10(3): e0118270.
[59]Wetmur JG, Kumar M, Zhang L, et al. Molecular haplotyping by linking emulsion PCR: analysis of paraoxonase 1 haplotypes and phenotypes. Nucleic Acids Res, 2005, 33(8): 2615–2619.
[60]Huang DW, Jiang MK, Zheng X, et al. Towards better precision medicine: PacBio single-molecule long reads resolve the interpretation of HIV drug resistant mutation profiles at explicit quasispecies (haplotype) level. J Data Mining Genomics Proteomics, 2016, 7(1): 182.
[61]Bansal V, Halpern AL, Axelrod N, et al. An MCMC algorithm for haplotype assembly from whole-genome sequence data. Genome Res, 2008, 18(8): 1336–1346.
[62]Bansal V, Bafna V. HapCUT: an efficient and accurate algorithm for the haplotype assembly problem.Bioinformatics, 2008, 24(16): 153–159.
[63]Lo C, Bashir A, Bansal V, et al. Strobe sequence design for haplotype assembly. BMC Bioinformatics, 2011,12(S1): S24.
[64]Edge P, Bafna V, Bansal V. HapCUT2: robust and accurate haplotype assembly for diverse sequencing technologies. Genome Res, 2017, 27(5): 801–812.
[65]Schiffels S, Durbin R. Inferring human population size and separation history from multiple genome sequences.Nat Genet, 2014, 46(8): 919–925.
[66]He G, Luo X, Tian F, et al. Haplotype variation in structure and expression of a gene cluster associated with a quantitative trait locus for improved yield in rice.Genome Res, 2006, 16(5): 618–626.
[67]He C, Fu J, Zhang J, et al. A gene-oriented haplotype comparison reveals recently selected genomic regions in temperate and tropical maize germplasm. PLoS ONE,2017, 12(1): e0169806.
[68]Nilsen KT, N’Diaye A, Maclachlan PR, et al. High density mapping and haplotype analysis of the major stem-solidness locus SSt1 in durum and common wheat.PLoS ONE, 2017, 12(4): e0175285.
[69]Lin X, Luo Y, Tang BH, et al. Study on hybrid induced haploid in maize. Shaanxi J Agr Sci, 2017, 63(2): 17–18(in Chinese).林霞, 罗勇, 唐秉晖, 等. 品种间杂交诱导玉米孤雌生殖单倍体的研究. 陕西农业科学, 2017, 63(2): 17–18.
[70]Liu ZJ, Shen LB, Zhu J, et al. The cloning and haplotype analysis of α-momorcharin from bitter gourd. Jiangsu J Agr Sci, 2017(5): 1117–1123 (in Chinese).刘子记, 申龙斌, 朱婕, 等. 苦瓜 α-苦瓜素基因克隆与单倍型分析. 江苏农业学报, 2017(5): 1117–1123.
[71]Welch KO, Marin RS, Pandya A, et al. Compound heterozygosity for dominant and recessive GJB2 mutations:effect on phenotype and review of the literature. Am J Med Genet A, 2007, 143A(14): 1567–1573.
[72]Fong CY, Mumford AD, Likeman MJ, et al. Cerebral palsy in siblings caused by compound heterozygous mutations in the gene encoding protein C. Dev Med Child Neurol, 2010, 52(5): 489–493.
[73]Shimizu H, Takizawa Y, Pulkkinen L, et al.Epidermolysis bullosa simplex associated with muscular dystrophy: phenotype-genotype correlations and review of the literature. J Am Acad Dermatol, 1999, 41(6):950–956.
[74]Drysdale CM, Mcgraw DW, Stack CB, et al. Complex promoter and coding region 2-adrenergic receptor haplotypes alter receptor expression and predictin vivoresponsiveness. Proc Natl Acad Sci USA, 2000, 97(19):10483–10488.
[75]Kitzman JO, Snyder MW, Ventura M, et al. Noninvasive whole-genome sequencing of a human fetus. Sci Transl Med, 2012, 4(137): 137ra76.
[76]Matias FI, Galli G, Correia Granato IS, et al. Genomic prediction of autogamous and allogamous plants by SNPs and haplotypes. Crop Sci, 2017, 57: 2951–2958.
[77]Schatz MC, Delcher AL, Salzberg SL. Assembly of large genomes using second-generation sequencing. Genome Res, 2010, 20(9): 1165–1173.
