地黄根部响应涝胁迫关键基因的鉴定△
2017-09-21王翠英李鑫宇王潇然李明杰张重义陈新建
王翠英,李鑫宇,王潇然,李明杰,张重义*,陈新建*
(1.河南农业大学 生命科学学院,河南 郑州 450002; 2.福建农林大学 中药材GAP研究所,福建 福州 350002)
·中药农业·
地黄根部响应涝胁迫关键基因的鉴定△
王翠英1,李鑫宇1,王潇然1,李明杰2,张重义2*,陈新建1*
(1.河南农业大学 生命科学学院,河南 郑州 450002; 2.福建农林大学 中药材GAP研究所,福建 福州 350002)
目的:本研究利用RNA-seq技术,对涝胁迫下的地黄根部差异表达基因进行了鉴定,为揭示地黄特异响应涝胁迫的分子机制奠定基础。方法:以温“85-5”怀地黄品种为供试材料,采用人工模拟涝胁迫的方法处理地黄植株,利用升级版数字基因表达谱技术分别对对照及处理样品进行测序,测序结果与转录组文库比对获得表达基因,利用RPKM方法计算基因的表达量,以FDR<0.005和|log2 ratio|≥1为标准,筛选差异表达基因,并对差异表达基因进行GO和KEGG注释和富集分析。结果:涝害胁迫处理后共筛选到7249差异表达基因,上调表达2505个(约35%),下调表达4744个(约65%)。结论:KEGGPathway显著性富集分析发现基础代谢、次生代谢、植物激素信号转导途径、淀粉和蔗糖代谢途基因包含的比例较多。功能分析发现,涝害条件下参与乙醇发酵、乙烯合成等途径的基因被显著上调,钙信号途径的基因异常,同时发现许多转录因子呈现差异表达,如WRKY、GRAS和NAC这些与胁迫相关的基因发生上调表达,而调控正常生长的转录因子如BHLH、MYC、BYB、MADS-box等则是以下调表达为主,由此证实涝害胁迫严重影响植物的正常生长发育。
地黄;升级版数字基因表达谱;人工模拟涝害胁迫;差异表达基因;转录因子
涝害是世界上许多国家面临的重大自然灾害之一[1]。涝害能扩大植物根际分子氧的运动速率而降低氧气供应导致植物缺氧或者低氧[2]。低氧能够引起能源和碳水化合物的危机,植物本身必须通过调节能量的消耗储备能量。低氧能引起一些基因表达的变化,这些基因与碳水化合物的分解、糖酵解、乙醇发酵途径、脂质代谢、乙烯合成、植物激素调解过程、钙信号、ROS产生[3]、转录因子等相关[4]。
地黄RehmanniaglutinosaL.玄参科地黄属多年生草本植物,是我国著名的“四大怀药”之一[5]。地黄块根膨大需要大量的氧气供应,要求土壤含水量约为10%~30%。地黄植株受到涝害胁迫以后会出现烂根现象,且很容易导致死亡,严重影响产量[6]。然而目前国内外对地黄涝害的研究尚未见报道。
本研究利用RNA-seq技术获得地黄涝胁迫下的基因表达谱数据,通过生物信息学方法筛选涝胁迫的差异表达基因,并对这些基因进行功能、代谢和调控途径的分析。利用qPCR对差异表达基因进行了验证。研究地黄在涝害胁迫下基因的差异表达,为进一步鉴定和涝胁迫响应的关键基因,揭示地黄对涝害敏感的分子机制奠定基础,为地黄抗涝害基因工程改良提供理论依据。
1 材料与方法
1.1 实验材料
本研究采用广泛种植且涝害反应最为明显的“温85-5”怀地黄品种为供试材料。在河南农业大学生命科学院分子生物学实验室和河南农业大学中药材研究所培养室中进行胁迫模拟试验(盆栽试验)。涝胁迫:出苗20 d选长势均一的材料进行处理,堵塞盆底部排水口,先进行10 d持续绕水最大持水量W的80%,稍停3 d后继续10 d持续绕水最大持水量W的90%,稍停3 d后堵塞盆底部排水口,每天浇灌最大持水量(W)使土表水保持2 mm高度直到所有植株出现有气生根生成;胁迫处理完成后,选取处理和对照的较为一致地黄整株根系,设置3个生物学重复,液氮速冻后,放入-80 ℃保存。
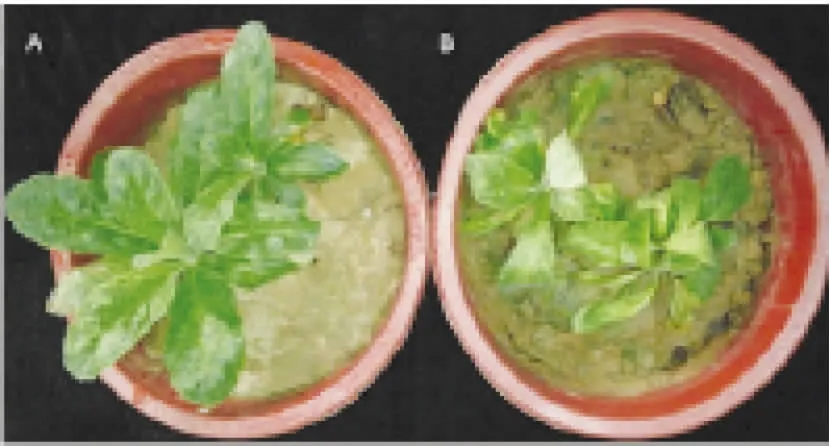
图1 A.对照; B.涝害处理
1.2 RNA提取和cDNA的合成
按TaKaRa TRIzol Reagent(Invitrogen)RNA试剂
盒说明书的要求分别提取对照和处理材料根部的总RNA,并通过琼脂糖凝胶电泳和Nanodrop2000来检测其纯度和提取的完整性,获得高质量RNA后,-80 ℃保存备用。
根据TaKaRa RevertAid TM First Strand cDNA Synthesis Kit试剂盒说明书进行操作把获得的高质量的RNA反转录为cDNA,放入-80 ℃保存,用于下一步qRT-PCR。
1.3 基因表达文库构建
提供浓度≥300 ng·μL-1、总量≥6 μg、OD260/280为1.8-2.2的对照和处理材料的地黄根部总RNA样品,由深圳华大基因科技有限公司对RNA样品进行后续处理。使用Illumina HiSeqTM 2000对质控合格文库完成测序。测序仪所得的原始图像数据经Base Calling转化为原始序列数据(raw reads),然后经过除杂质数据(去除原始序列中含adaptor序列、N的比例大于10%的序列、质量值Q≤5的碱基数占整个read的50%以上的低质量序列)获得高质量干净序列(Clean Reads),以FASTQ文件格式存储。使用短序列比对软件SOAPaligner/SOAP2[7]将Clean Reads分别比对到参考基因序列上(允许2个碱基错配)。参考序列为课题组前期获得的两个6G和20G的地黄转录组数据库(http://www.ncbi.nlm.nih.gov/sra,注册号SRX269425和SRX269426)合并拼接后构建的地黄转录组文库。利用唯一比对上的基因的reads数目和比对上的参考序列的总reads数计算基因的表达量,计算使用RPKM(Reads Per Kb per Million mapped reads)方法对测序深度和基因长度进行归一化,如果一个基因存在多个转录本,则用该基因的最长转录本计算其测序覆盖度和表达量[8]。
1.4 差异基因筛选和功能注释
参照Audic等[9]基于测序的差异表达基因的检测方法,筛选两样本间的差异表达基因。然后对差异检验的p-value作多重假设检验校正,通过控制FDR(False Discovery Rate)来决定p-value的域值。即FDR≤0.001且∣log2Ratio∣≥1(倍数差异不低于2倍)的基因为差异表达基因[10]。后续分析均基于差异表达基因。Gene Ontology功能显著性富集分析采用Gene Ontology(GO)数据库(http://www.geneontology.org/),使用 Blast2GO 软件(默认参数)将差异表达基因向数据库的各个term映射,从而获得该基因的GO功能分类注释,计算每个term的基因数目。以通过Bonferroni校正之后的差异检验值p-value≤0.05为阈值,应用超几何检验,找出与整个背景相比,在差异表达基因中显著富集的GO term。通过GO功能显著性富集分析确定差异表达基因所行使的主要生物学功能。Pathway显著性富集分析采用 KEGG 数据库(kyoto encyclopedia of genes and genomes),将差异表达基因向数据库映射,并且对pathway进行显著性富集分析,统计各个pathway中基因数目及富集程度,分析方法与GO富集分析方法相同[11]。通过Pathway显著性富集确定差异表达基因最主要参与的信号转导途径和生化代谢途径。
1.5 实时荧光定量PCR分析
利用qRT-PCR技术验证高通量测序数据的可靠性。选取上、下调表达基因各5个差异表达基因,根据转录组基因序列,利用primer premier 5.0软件设计荧光定量引物(表1),扩增产物长度为150~300 bp。以18srRNA为定量分析的内参基因。取对照和涝胁迫处理材料的cDNA样品经内参基因PCR检测合格后用于qRT-PCR的cDNA模板,应用全式金荧光定量PCR试剂盒2×TransStart Green qPCR SuperMix,Passive Reference Dye的反应体系,采用Bio-Rad公司生产的荧光定量PCR仪进行扩增反应。扩增程序为95 ℃预变性3 min;95 ℃ 30 s、59 ℃ 30 s、72 ℃ 30 s,共40个循环;95 ℃ 1 min,60 ℃ 1 min。取3个平行孔校正后使用2-△△Ct法计算相对表达量[12]。

表1 qRT-PCR引物
2 结果与分析
2.1 测序数据质量评估和基因覆盖度分析
经Illumina HiSeqTM 2000测序,对照组和涝胁迫处理组分别获得了281、603、980和301、565、012个总碱基对(Total Base Pairs),经过去除杂质处理后,对照组和处理组得到的clean reads数分别为5、747、020条(占97.81%)和6、154、388条(占96.74%),去杂后获得的clean reads比例接近, 说明文库构建和测序质量较高。在缺乏基因组的遗传背景和注释信息量的限制下,本实验比对到转录组的总序列数(Total Mapped Reads)分别有4、683、336和4、964、331条(占81.49%和80.66%)。其中完全比对上的基因分别有3、273、007和3、437、381条(占56.95%和55.85%),特异匹配的分别有2、943、032和2、993、723条(占51.21%和48.64%)(表2),能够较好地反应基因表达的真实
情况[13],Clean Reads中覆盖度达90%以上的分别占7%和6%(图2),说明上述样品制备和测序质量良好。测序饱和度分析表明,两组样品所检测到的基因数都已趋于饱和。此外,测序随机性分析显示,reads在基因各部位分布得较为均一,说明序列分析过程中mRNA没有降解和偏向性的影响,可以用于后续数据分析。
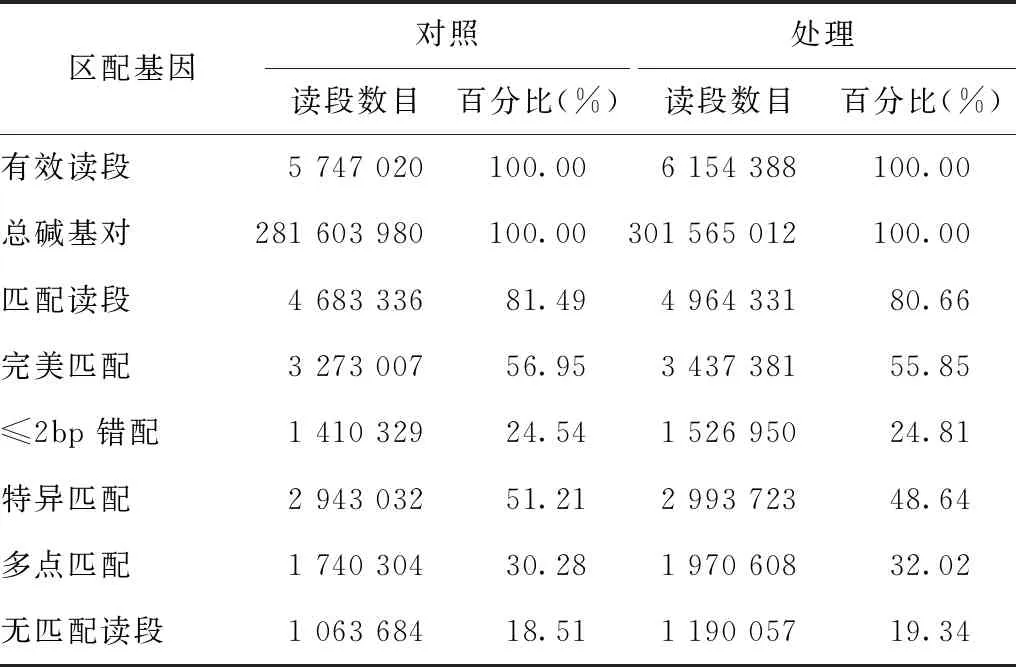
表2 DGE测序数据统计结果
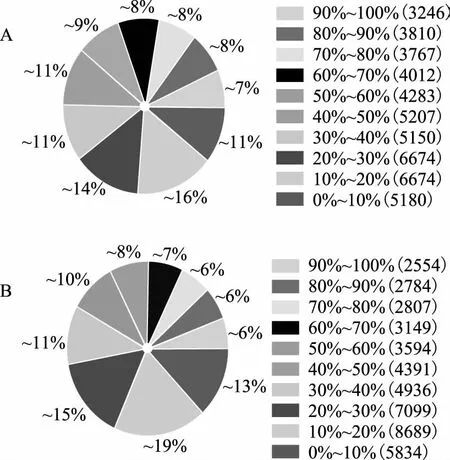
注:A.对照组;B.处理。图2 基因覆盖度分布
2.2 差异表达基因的筛选
根据数字基因表达谱差异基因检测方法,以RPKM计算基因表达量,在FDR<0.005和|log2ratio(处理/对照)|≥1范围内筛选差异表达基因(图3)。经过严格筛选最后确定涝害胁迫处理后有7249个显著差异表达基因。其中,上调的有2505个,其中表达量差异大于1000倍(|log2ratio|≥10)以上的基因有149个,上调倍数最高为86 939;下调的有4744个,其中表达量差异大于1000倍(|log2ratio|≥10)以上的基因有142个,下调倍数最高为22 906。大量基因的差异表达很明显的说明,地黄在涝害胁迫下很可能进行着活跃的基因表达活动,但基因表达差异高度不均一,表达水平变化主要集中在16倍以内,表达量差异较大的基因数目较少,且大多数基因表现出下调表达。
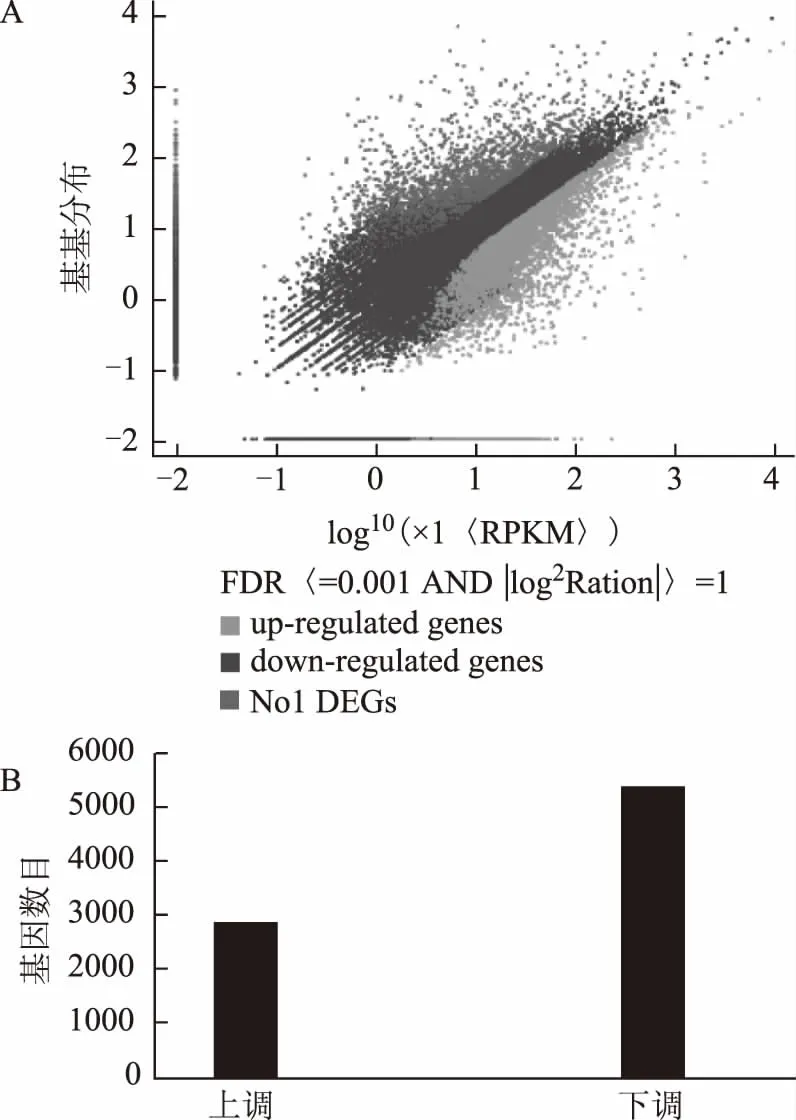
图3 差异表达基因的分布和数目
2.3 差异表达基因的Gene Ontology功能显著性富集分析
利用GO分析方法对差异基因进行分析,通过与转录组比对得到已注释差异表达基因共有5169个,在从数据库中查询到校正P≤1.0的3890个GO本体信息条目中,基因的分子功能(molecular function)、所处的细胞位置(cellular component)、参与的生物过程(biological process)所占比例分别为27.94%、8.76%、63.3%。以校正P≤0.05 为显著性富集标准,对差异表达基因GO注释分类条目进行显著性富集分析。结果表明,其参与的生物学过程主要包括细胞加工(cellular process)、代谢过程(metabolic process)、刺激响应(response to stimulus)、生物调节(biological regulation)、生物过程的监管(regulation of biological process)、发育过程(developmental process)以及生长过程(growth)等(图4)。说明涝胁迫影响到地黄体内的许多生理生化过程。
2.4 差异表达基因的Pathway分析
在生物体内,基因通过相互作用网络来行使生物学功能,利用KEGG数据库中对差异表达基因进行分析,通过显著性富集来确定这些基因主要参与的生化代谢途径和信号转导途径,有利于进一步了
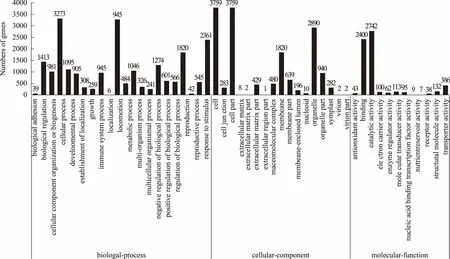
图4 差异表达基因GO功能分类
解这些基因的生物学功能。结果表明,涝胁迫下地黄差异表达基因有4276个可以被KEGG数据库注释,这些基因分布在126条pathway中。以P≤0.05为条件富集得到30条差异表达基因中显著性富集的通路(pathway)。这些显著性富集大致可以分成六类:(1)次生代谢;(2)激素代谢和信号转导途径;(3)氨基酸代谢;(4)光合作用;(5)脂类代谢;(6)转运体代谢。在能够注释到这些路径图中的基因中,氧化磷酸化、光合作用和细胞活动相关基因所占比例相对较低;与次生代谢相关基因占到54.5%。
2.5 数字基因表达谱的验证
为了检验升级版数字基因表达谱数据的可靠性,对挑选基因进行qRT-PCR分析。检验以18 s为内参基因。结果显示涝胁迫下这些基因表达的变化上下调趋势一致(图5)。说明表达谱数据可靠。
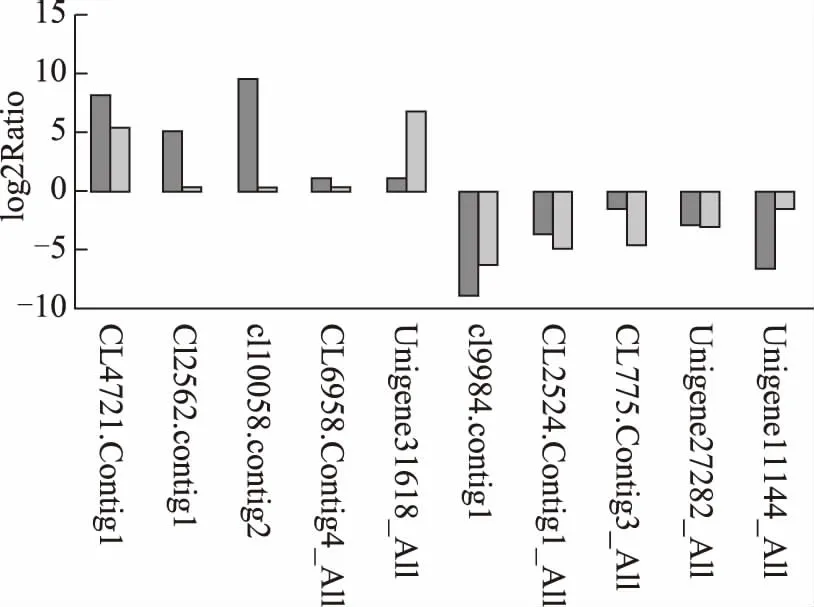
图5 差异表达基因的qRT-PCR验证
3 讨论
涝害是重要的自然灾害之一,涝害引起的植物体内基因的差异表达研究是植物研究热点之一,近年来在拟南芥[14]、水稻[15]、番茄[16]、猕猴桃[17]等均匀涝胁迫中的基因差异表达的研究报道。作为收获块根的地黄,由于涉及到根系膨大,对水分更为敏感,研究涝胁迫下根部基因表达差异,显得更为紧迫和重要。同时本研究结果也为收获块根类作为提供有益的借鉴。在基因组信息匮乏的情况下,作者利用转录组和数字表达谱技术,得到了一套涝胁迫下地黄根部差异表达基因数据,分析这些差异表达的基因及其功能,使我们对地黄涝害有了更深入和全面的了解。
3.1 乙醇发酵是涝害的前期事件
涝害对植物的影响主要是缺氧或低氧,植物在低氧条件下激活乙醇发酵,丙酮酸在丙酮酸脱羧酶作用下生成乙醛,然后由乙醇脱氢酶还原成乙醇。在本研究中发现与乙醇发酵相关的丙酮酸脱羧酶有5个上调变化,乙醇脱氢酶则以下调为主。表明在地黄涝害中乙醇发酵途径被激活,同时分解乙醇相关酶的编码基因呈下调表达。然而,大豆的根中丙酮酸脱酸酶和乙醇脱氢酶呈上调变化[18],在猕猴桃中乙醇脱氢酶和丙酮酸脱酸酶基因呈显著上调变化[17]。推测可能由于地黄为块根植物,生长发育过程显著区别于其他直根系作物,根系对响应涝胁迫机制也存在一定差异。
3.2 Ca信号系统紊乱的放大了涝害的影响
Ca2+作为植物细胞中的第二信使,越来越多的研究表明钙离子对各种逆境的信号转导起重要作用。植物在非生物逆境胁迫下,钙离子的浓度会发生特异的变化,诱导钙信号,钙信号通过下游的钙结合蛋白进行感受和转导,进而引起细胞内一系列的反应抵御逆境胁迫。在拟南芥、水稻、玉米、小麦中发现低氧能引起Ca2+快速提高,激活钙信号的转导[19]。研究表明Ca2+信号的转导与运输与钙依赖蛋白激酶和钙调素有关,由钙依赖蛋白完成信号传递,再由H+/Ca2+和Ca2+-ATP等反转运系统来终结逆境信号[20]。本研究发现钙依赖蛋白激酶2个下调表明钙信号向下游传递受到影响,H+/Ca2+3个下调和Ca2+-ATP 7个下调和6个上调表明钙信号终结逆境信号出现紊乱,表明Ca2+信号对地黄涝害信号的传递与运输起到重要的作用。
3.3 乙烯的产生使植株地上部表现出伤害症状
涝害影响植物内源激素的合成与运输。涝害条件下,植物体内乙烯含量增多[21],诱导根中ACC(1-氨基环丙烷1-羧酸)合成基因促进根中ACC的合成。ACC随蒸腾叶流由根系向地上部分运输,地上部分的ACC在通气条件下转化成乙烯。本研究中发现根部ACC合成酶(乙烯合成的关键酶)呈极显著的上调变化,表明涝害条件下根部合成了ACC。在根部没有检出ACC氧化酶的表达差异,说明ACC氧化成乙烯的过程不在根部发生,而是在地上部。乙烯在负调控地黄的生长发育,使植株地上部表现出萎焉症状。
3.4 转录因子的错乱表达加剧了涝害的程度
转录因子是基因表达中一类重要的调控因子,通过与目标基因启动子中特定的DNA序列即顺式作用元件结合,激活或抑制靶基因的转录表达,研究表明植物转录因子能调控植物胁迫反应中特异基因的表达[22]。在本研究中有12个转录因子家族被发现参与地黄的涝害(如表3),其中WRKY家族有36个上调,18个下调;BHLH家族有10个上调,28个下调;MYC2家族有7个上调,22个下调;MYB家族有7个上调和13个下调等,表明这些转录因子在地黄涝害过程中对调控基因表达起着重要作用。GRAS转录因子是植物特有的转录因子,已有报道表明其参与植物的生物胁迫和非生物胁迫相关的应答过程[23]。目前已经在拟南芥[24]、水稻[25]、葡萄[26]、番茄[27]等多种植物中发现GRAS家族基因参与作物的胁迫应答。本研究中在地黄中发现有4个GRAS转录因子,其中3个上调和1个下调,表明GRAS转录因子可能参与地黄涝害的应答过程。NAC转录因子也是植物特有的一类转录因子,已有研究表明NAC转录因子在植物抗逆反应中起重要调控作用[28]。本研究中发现有4个NAC转录因子,3个上调和1个下调,表明NAC转录因子可能参与地黄涝害的调控。
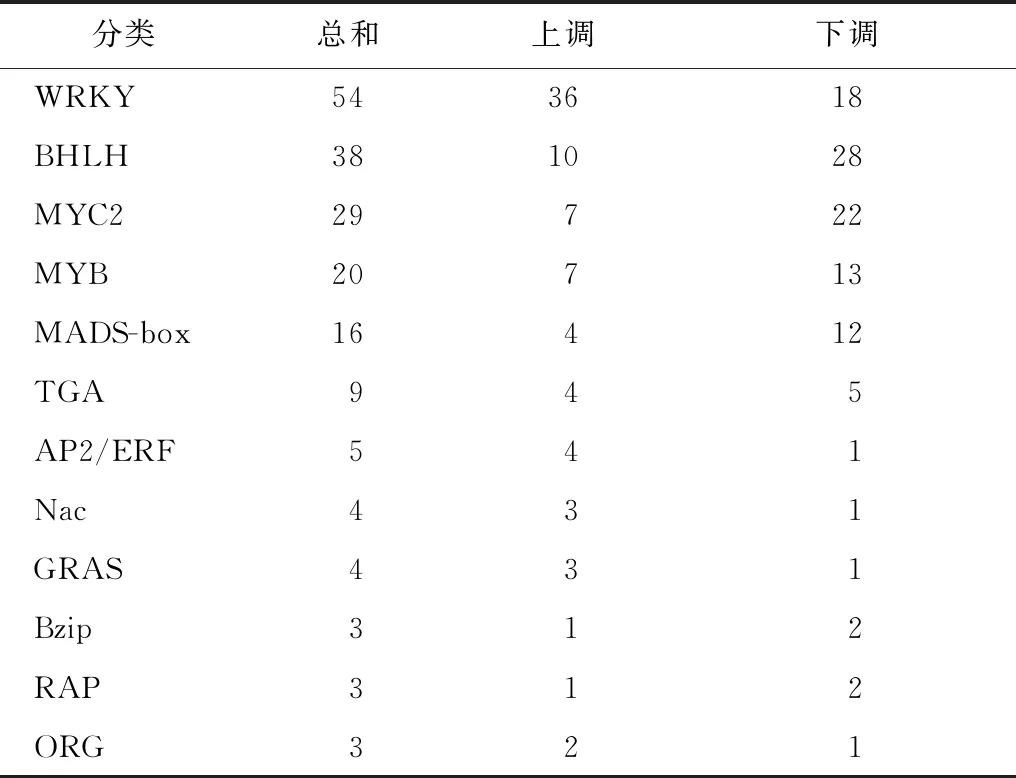
表3 差异表达转录因子的分布
3.5 关键基因的下调表达使得淹水植株生长受阻
在淹水条件下地黄根部许多控制生长发育的关键基因被下调表达,使得淹水植株生长受阻。如细胞周期素(cyclin),是控制细胞分裂的关键蛋白。在检测到的41个cycling的基因中有39下表达。细胞骨架蛋白myosin,与细胞的分裂、细胞的形状有着密切的关系。在根部检测到25个myosin基因,有24个为下调表达。淹水条件下根部分裂受阻,症状萎焉,与这些关键基因的下调表达有关。地黄根部淹水条件下的这些发现,在其他植物中尚未见报道。
4 结论
本研究通过数字表达谱升级版技术,在缺乏基因组,遗传背景不是十分清楚的情况下,对照组和涝害处理组得到的Clean Reads数分别为5、747、020条和6、154、388条,通过差异表达基因筛选以及功能注释,从中共筛选到7249个显著差异表达基因,有2505(约35%)个基因出现上调表达,有4744(约65%)个下调表达的基因。为进一步研究地黄涝胁迫网络和分子机制提供理论依据。
[1] 蒋薇,刘登望,李林.作物涝害研究进展[J].南方农业学报,2010,41(5):432-435.
[2] Christianson J A,Llewellyn D J,Dennis E S,et al.Global gene expression responses to waterlogging in roots and leaves of cotton(GossypiumhirsutumL.)[J].Plant Cell Physiol ,2009,51(1):21-37.
[3] Liu F,Vantoai T,Moy L P,et al.Global transcription profiling reveals novel insights into hypoxic response in Arabidopsis[J].Plant Physiol,2005,137(3):1115-29.
[4] Lee Y H,Kim K S,Jang Y S,et al.Global gene expression responses to waterlogging in leaves of rape seedlings[J].Plant Cell Reports,2014,33(2):289-99.
[5] 温学森,杨世林,魏建和,等.地黄栽培历史及其品种考证[J].中草药,2002,33(10):946-949.
[6] 杜韧强,张娟,康尔艳.地黄栽培技术[J].吉林农业,2005(1):20-21.
[7] Chen S,Zhang C,Chen F,et al.METHOD FOR DETECTING GENETIC VARIATION:WO,EP 2772549 A4[P].2015.
[8] Li R,Yu C,Li Y,et al.SOAP2:an improved ultrafast tool for short read alignment[J].Bioinformatics,2009,25(15):1966-7.
[9] Audic S,Claverie J M.The significance of digital gene expression profiles[J].Genome Res,1997,7(10):986-995.
[10]Bennett S.Solexa Ltd[J].Pharmacogenomics,2004,5:433-438.
[11]Minoru K,Michihiro A,Susumu G,et al.KEGG for linking genomes to life and the environment[J].Nucleic Acids Res,2015,36:53-72.
[12]Livak K J,Schmittgen T D.Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T))Method[J].Methods,2001,25(4).
[13]Govind G,Harshavardhan V T,Patricia J K,et al.Identification and functional validation of a unique set of drought induced genes preferentially expressed in response to gradual water stress in peanut[J].Molecular Genetics and Genomics,2009,281(6):591-605.
[14]Narsai R,Whelan J.How unique is the low oxygen response? An analysis of the anaerobic response during germination and comparison with abiotic stress in rice and Arabidopsis[J].Powder Diffr,2014,29(S1):102-S106.
[15]Lasanthi-Kudahettige R,Magneschi L,Loreti E,et al.Transcript profiling of the anoxic rice coleoptile[J].Plant Physiol,2007,144(1):218-231.
[16]Shiono K,Takahashi H,Colmer T D,et al.Role of ethylene in acclimations to promote oxygen transport in roots of plants in waterlogged soils[J].Plant Sci,2008,175(1-2):52-58.
[17]Tougou M,Hashiguchi A,Yukawa K,et al.Responses to flooding stress in soybean seedlings with the alcohol dehydrogenase transgene[J].Plant Biotechnol,2012,29(3):301-305.
[18]Zhang J Y,Huang S N,Mo Z H,et al.De novo transcriptome sequencing and comparative analysis of differentially expressed genes in kiwifruit under waterlogging stress[J].Mol Breeding,2015,35(11):1-12.
[19]Wang F,Chen Z,Liu X,et al.Tissue-specific root ion profiling reveals essential roles of the CAX and ACA calcium transport systems for hypoxia response in Arabidopsis[J].J Exp Bot,2016.
[20]张和臣,尹伟伦,夏新莉.非生物逆境胁迫下植物钙信号转导的分子机制[J].植物学报,2007,24(1):114-121.
[21]樊明寿,张福锁.植物通气组织的形成过程和生理生态学意义[J].植物生理学报,2002,38(6):615-618.
[22]陈儒钢,巩振辉,逯明辉,等.植物抗逆反应中的转录因子网络研究进展[J].农业生物技术学报,2010,18(1):126-134.
[23]李桂英,田玉富,杨成君.植物GRAS家族转录因子的研究现状[J].安徽农业科学,2014(14):4207-4210.
[24]郭华军,焦远年,邸超,等.拟南芥转录因子GRAS家族基因群响应渗透和干旱胁迫的初步探索[J].植物学报,2009,44(3):290-299.
[25]OsGRAS23,a rice GRAS transcription factor gene,is involved in drought stress response through regulating expression of stress-responsive genes[J].BMC Plant Bio,2015,15(1):1-13.
[26]孙欣,王晨,房经贵,等.葡萄GRAS基因家族生物信息学分析[J].江西农业学报,2011,23(7):1-8.
[27]Huang W,Xian Z,Xia K,et al.Genome-wide identification,phylogeny and expression analysis of GRAS gene family in tomato[J].BMC Plant Bio,2015,15(1):1-18.
[28]孙利军,李大勇,张慧娟,等.NAC转录因子在植物抗病和抗非生物胁迫反应中的作用[J].遗传,2012,34(8):993-100.
IdentificationofKeyGenesinResponsetoWaterloggingStressinRootofRehmanniaglutinosa
WANGCuiying1,LIXinyu1,WANGXiaoran1,LIMingjie2,ZHANGZhongyi2,CHENXinjian1*
(1.SchoolofLifeSciences,HenanAgriculturalUniversity,Zhengzhou450002,China; 2.InstituteofChineseHerbalMedicineGoodAgriculturePractice,FujianAgricultureandForestryUniversity,Fuzhou350002,China)
Objective:We detected differentially expressed genes ofRehmanniaglutinosatuberous root under submergence stress using RNA-seq technique,which would lay foundation on exploring molecular mechanisms of specific response to waterlogging stress forR.glutinosa.Methods:Wen“85-5” was used as material in this study,of which transcriptome was investigated under submergence stress using upgraded digital gene expression profiling(DGE)technique.The expression level was normalized based on Reads Per Kilobase per Million mapped reads(RPKM)method.The differentially expressed genes were selected using |log2 Ratio|≥1 and FDR adjustedp-value of <0.001 as cutoff,which were performed GO and KEGG pathway analysis.Results:A total of 7249 genes,with 2505 up-regulated(35%)and 4744 down-regulated(65%),were shown different expression after submergence stress.Conclusion:The KEGG enrichment analysis of these differentially expressed genes revealed that numerous genes mapped to “basic metabolism”,“secondary metabolism”,“hormone signaling pathways”,and “starch and sugar metabolism pathways”.The functional analysis suggested that some genes involved in ethanol fermentation pathways and ethylene biosynthesis showed significant up-regulation,and the genes associated with calcium signaling pathways presented express perturbation.In addition,plenty of genes of transcription factors showed differential expression,e.g.,WRKY,GRAS and NAC related to stress presented up-regulation.However,transcription factors modulated development,e.g.,BHLH,MYC,BYB and MADS-box,mainly disclosed down-regulation.It was indicated that submergence stress was severely harmful to plants.
Rehmanniaglutinosa;upgraded-DGE;artificial submergence stress;differentially expressed genes;transcription factors
国家自然科学基金项目(30600209)
] 张重义,教授,研究方向:中药资源生态学、药用植物优质栽培,Tel:(0591)83742793,E-mail:hauzzy@163.com;陈新建,教授,研究方向:地黄连作障碍分子机制研究,Tel:(0371)63558722,E-mail:xinjianl@yahoo.com
10.13313/j.issn.1673-4890.2017.2.015
2016-05-10)
*[
