通用引物扩增甲型流感病毒基因组及其高通量测序
2017-01-14王楷宬纪海旺庄青叶王素春于建敏许荣强陈继明
王楷宬,邱 源,纪海旺,彭 程,庄青叶,王 通,王素春,于建敏,许荣强,陈继明
(1.中国动物卫生与流行病学中心,山东青岛 266032;2. 崂山区王哥庄街道农业服务中心,山东青岛 266105)
通用引物扩增甲型流感病毒基因组及其高通量测序
王楷宬1,邱 源1,纪海旺2,彭 程1,庄青叶1,王 通1,王素春1,于建敏1,许荣强1,陈继明1
(1.中国动物卫生与流行病学中心,山东青岛 266032;2. 崂山区王哥庄街道农业服务中心,山东青岛 266105)
为探索使用高通量测序方法测定甲型流感病毒的基因组,设计了一对通用引物进行甲型流感病毒全基因组的RT-PCR扩增,并分析了该方法的敏感性。结果显示,该方法能够扩增的甲型流感病毒多种亚型的全部8个节段的基因,其扩增敏感性为100 pg RNA。
甲型流感病毒;通用引物;基因组;高通量测序
甲型流感病毒属于正黏病毒科流感病毒属,为有包膜分节段的单负链RNA 病毒。其基因组由8个单股负链RNA片段组成,每个基因在3´末端和5´末端都带有12~13个保守核苷酸序列。这8个片段可编码10种蛋白,其中8个蛋白(HA、NA、PB1、PB2、NP、M1、M2和PA)为结构蛋白,NS1和NS2为非结构蛋白,位于宿主细胞的胞质中[1]。根据HA 和NA 抗原性的差异,甲型流感病毒可分为若干亚型,迄今已发现有18种HA亚型(H1~H18)和11种NA亚型(N1~N11)[2-4]。任何一对HA 和NA 均可组合成一个亚型,如H1N1、H5N1、H7N9等。
甲型流感病毒能产生低保真RNA聚合酶。其可引起流感病毒的高突变率以及基因重组,可使流感病毒呈现分子多样性,使每个病毒亚型可进化为多个分支[5]。由于甲型流感病毒亚型多、突变率高、易发生重配,因此对流行毒株进行基因组分析尤为重要。随着新一代测序技术的发展,采用高通量测序方法进行甲型流感病毒基因组的测定[6],已被科研人员广泛接受。已经有研究人员报道,在1个RT-PCR反应中,使用1对通用引物扩增甲型流感病毒所有8个基因节段的方法,从而使得流感病毒全基因组的测序更为简单[7]。传统测序方法虽然经济高效,但仅能测定某一特异性扩增片段,且不能测定准种[8]。而我国现在用于高通量测序平台的甲型流感病毒基因组扩增试剂主要依赖进口,购买该试剂的费用较高。本文参照文献,设计了1对通用引物进行甲型流感病毒全基因的RT-PCR扩增,并分析了该方法的敏感性。
1 材料和方法
1.1 材料和仪器
Ion Xpress Plus Fragment Library Kit、Ion PGM Template OT2 200 Kit、Ion PGM Sequencing 200 Kit V2、E-Gel SizeSelect 2% Agarose、Ion 318 Chip Kit V2、 Dynabeads MyOne Streptavidin C1 Beads、Ion Xpress Batcode Aaptors、Qubit核酸浓度测定仪及配套试剂,均购自Life technologies公司;超净工作台(Forma Scientifc);移液器(Eppendorf);孵化器(德州诚信孵化设备有限公司);高速台式离心机(Heraeus Biofuge primoR);PCR扩增仪(Perkin Elmeter Gen Amp PCR System 9600)。
1.2 毒株和核酸提取
各亚型甲型流感毒株由中国动物卫生与流行病学中心禽病监测室保存,毒株详细信息见表1。病毒株接种9~11日龄鸡胚,72小时内收取鸡胚尿囊液进行病毒核酸RNA提取。按照说明书操作,使用RNA提取试剂盒(QIAamp Viral RNA Mini Kit,Qiagen)提取病毒RNA,并使用Qubit核酸浓度测定仪及配套试剂测定RNA浓度。
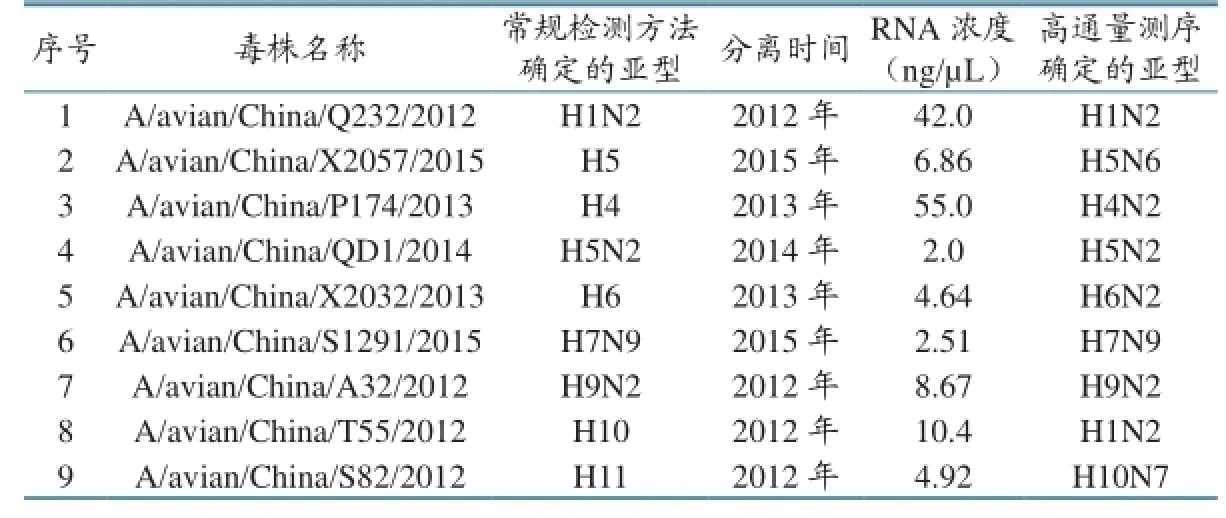
表1 禽流感病毒株信息
1.3 引物合成
按 照 文 献[6]合 成 引 物。MBTuni-12:5'-ACGCGTGATCAGCAAAAGCAGG;MBTuni-13:5'-ACGCGTGATCAGTAGAAACAAGG。
1.4 甲型流感病毒基因组扩增
使 用 PrimeScript™ One Step RT-PCR Kit Ver.2(Takara)进行甲型流感病毒全基因扩增,反应体系:2×1 Step Buffer 25 µL,MBTuni-12(10 pM)和MBTuni-13(10 pM)各2 µL,PrimeScript 1 Step Enzyme Mix 2 µL,RNA 10 µL,RNase Free dH2O 9 µL。反应条件 :45 ℃反转录1 h;95 ℃4 min;95 ℃变性15 s,60 ℃退火30 s,72 ℃延伸2 min,共40个循环;72 ℃延伸7 min。扩增产物经毛细管电泳进行鉴定。
1.5 文库构建
使用AMPure XP核酸纯化磁珠(Beckman)对扩增得到的甲型流感病毒基因组进行扩增,将纯化后的全基因cDNA稀释成35 µL中含有200 ng DNA的溶液,使用Ion Xpress Plus Fragment Library Kit将病毒全基因组DNA打断为200 bp的片段,加入含有Barcode的接头,构建为PGM测序仪可识别的DNA文库。
1.6 测序
将DNA文库稀释为26 pM,应用Ion PGM Template OT2 200 Kit对DNA文库进行测序前的样品处理。将处理后的样品加样至Ion 318 芯片,置于PGM测序仪进行测序。将测序序列经PGM自带的FluAnalysis(v4.0)软件进行序列拼接。
1.7 敏感性试验
以A/avian/China/A32/2012的基因组RNA为模板,稀释成含有1 ng/µL、100 pg/µL、10 pg/µL、1 pg/µL、0.1 pg/µL和0.01 pg/ µL的RNA溶液,使用PrimeScript™One Step RT-PCR Kit Ver.2) 和 引 物MBTuni-12/ MBTuni-13进行甲型流感病毒的全基因扩增,扩增产物经毛细管电泳进行鉴定。
2 结果与分析
2.1 流感病毒全基因组扩增及文库构建
使用该对引物能够扩增出9个毒株(8个HA亚型和3个NA亚型)的目的条带,得到该病毒全基因的所有节段扩增产物(图1)。纯化后的cDNA浓度均能满足测序文库构建的需要,经稀释后构建成含有Barcode接头的200 bp DNA文库。

图1 甲型流感病毒基因组扩增结果
2.2 测序数据分析
测序数据分析发现,该方法能够完成这些亚型的全基因组测序,测序数据拼接后可覆盖甲型流感病毒的所有8个节段。各毒株每个节段的平均测序深度见表2。包括各节段的5´和3´端序列,所有毒株的节段至少平均被测199次(A/avian/China/P174/2013的PA基因)。综合分析所有9个毒株8个节段的平均测序深度发现:M基因的平均测序深度最大,为5 533;PA基因的平均测序深度最小,为1 497。通过FluAnalysis(v4.0)软件对各毒株的HA和NA亚型进行分析,发现除第8和第9株通过高通量测序鉴定的亚型与常规检测结果不同外,其余均一致(表2)。
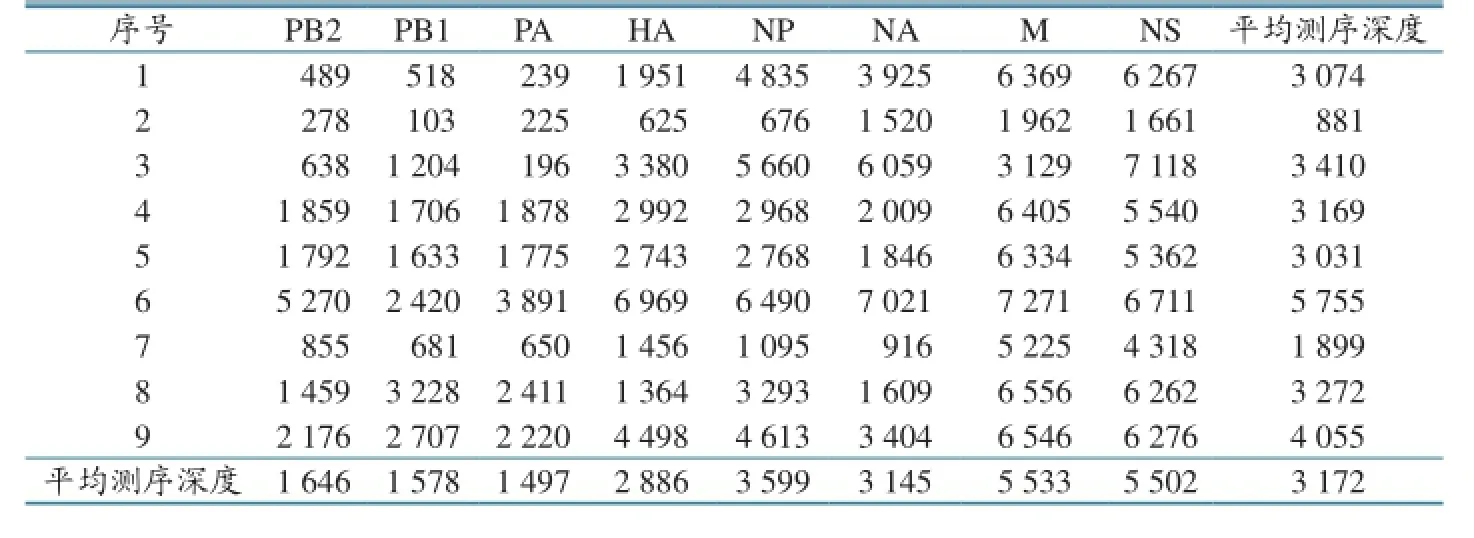
表2 毒株各节段的测序深度
2.3 敏感性试验结果
不同浓度的A/avian/China/A32/2012的基因组RNA,经使用PrimeScript™ One Step RT-PCR Kit Ver.2和引物MBTuni-12/ MBTuni-13进行甲型流感病毒全基因扩增。扩增产物毛细管电泳结果见图2。由图2可见,反应体系中加入10 µL浓度为10 pg/µL的RNA(100 pg)仍能检测到清晰的所有流感病毒节段的条带。

图2 不同浓度的引物RT-PCR检测甲型流感病毒敏感性的比较
3 讨论
甲型流感病毒全基因测序对了解流感病毒的分子生物学特性、基因重配机制、准种分析[14]等具有十分重要的意义。甲型流感病毒全基因测定的传统方法常是采用每个基因的特异性引物扩增的方法,扩增产物经直接测序或克隆测序得到全基因序列。这种方法操作繁琐,且针对某些亚型的甲型流感病毒需要采用亚型特异性引物进行扩增。这在毒株亚型未知的情况下,更会加大工作量。为了解决这些问题,使用通用的一对引物,一步RT-PCR扩增全部亚型的全基因方法被逐步建立和应用[1,9],但将全部8个节段的扩增产物在一个反应体系中进行测序仍是一项复杂工作。随着高通量测序的发展,这种混合样品的测序问题被逐步得到解决,从而使得甲型流感病毒的全基因测序技术得到进一步发展[6]。采用高通量测序,可以开展甲型流感病毒的监测[10]、亚型鉴定[11]、全基因测序与进化分析[12],以及突变频率[13]和准种分析[14]等工作。
我国用于甲型流感病毒全基因高通量测序的配套试剂全部依赖进口,国内鲜见有关通用引物扩增甲型流感病毒全基因的报道。本文依据文献报道,建立了此种通用引物扩增方法。结果显示,所采用的建库与测序方法能够测定甲型流感病毒的全基因,并且测序深度较大,能满足序列分析的需要。本实验在一次测序中完成了9种亚型甲型流感病毒的全部8个基因的测序,并分析出各毒株的HA和NA亚型。但常规测序判定为H10和H11的两株病毒,在本实验中却被分别分析为H1和H10,此结果产生的原因将会在下一步的研究中分析。此项技术的建立,将提高我国甲型流感病毒监测和分析的能力和效率,减少我国相关试剂的购买费用,待技术成熟后,可向科研院所和疾病防控机构推广。
[1] HOFFMANN E,STECH J,GUAN Y,et al. Universal primer set for the full-length amplification of all influenza A viruses [J]. Arch Virol,2001,146(12):2275-2289.
[2] FREIDL GS,BINGER T,MULLER MA,et al. Serological evidence of influenza a viruses in frugivorous bats from Africa [J]. PLoS One,2015,10(5):e0127035.
[3] TONG S,LI Y,RIVAILLER P,et al. A distinct lineage of infuenza A virus from bats [J]. Proc Natl Acad Sci U S A,2012,109(11):4269-4274.
[4] TONG S,ZHU X,LI Y,et al. New world bats harbor diverse infuenza A viruses [J]. PLoS Pathog,2013,9(10):e1003657.
[5] GHEDIN E,SENGAMALAY NA,SHUMWAY M,et al. Large-scale sequencing of human infuenza reveals the dynamic nature of viral genome evolution [J]. Nature,2005,437(7062):1162-1166.
[6] KAMPMANN ML,FORDYCE SL,AVILA-ARCOS MC,et al. A simple method for the parallel deep sequencing of full infuenza A genomes [J]. J Virol Methods,2011,178(1/2):243-248.
[7] ZHOU B,DONNELLY ME,SCHOLES DT,et al. Singlereaction genomic amplifcation accelerates sequencing and vaccine production for classical and Swine origin human infuenza a viruses [J]. J Virol,2009,83(19):10309-10313.
[8] DOMINGO E,BARANOWSKI E,RUIZ-JARABO CM,et al. Quasispecies structure and persistence of RNA viruses [J]. Emerg Infect Dis,1998,4(4):521-527.
[9] INOUE E,WANG X,OSAWA Y,et al. Full genomic amplifcation and subtyping of infuenza A virus using a single set of universal primers [J]. Microbiol Immunol,2010,54(3):129-134.
[10] MCGINNIS J,LAPLANTE J,SHUDT M,et al. Next generation sequencing for whole genome analysis and surveillance of infuenza A viruses [J]. J Clin Virol,2016,79:44-50.
[11] LIN Z,FAROOQUI A,LI G,et al. Next-generation sequencing and bioinformatic approaches to detect and analyze infuenza virus in ferrets [J]. J Infect Dev Ctries,2014,8(4):498-509.
[12] HOECKE S V D,VERHELST J,VUYLSTEKE M,et al. Analysis of the genetic diversity of infuenza A viruses using next-generation DNA sequencing [J]. BMC Genomics,2015,16:79.
[13] LI X,SHI J,GUO J,et al. Genetics,receptor binding property,and transmissibility in mammals of naturally isolated H9N2 Avian Infuenza viruses [J]. PLoS Pathog,2014,10(11):e1004508.
[14] KURODA M,KATANO H,NAKAJIMA N,et al. Characterization of quasispecies of pandemic 2009 infuenza A virus(A/H1N1/2009) by de novo sequencing using a nextgeneration DNA sequencer [J]. PLoS One,2010,5(4):e10256.
[15] GAO R,CAO B,HU Y,et al. Human infection with a novel avian-origin influenza A(H7N9) virus [J]. N Engl J Med,2013,368(20):1888-1897.
[16] XIONG C,ZHANG Z,JIANG Q,et al. Evolutionary characteristics of A/Hangzhou/1/2013 and source of avian influenza virus H7N9 subtype in China [J]. Clin Infect Dis,2013,57(4):622-624.
(责任编辑:朱迪国)
Genome Amplification of Influenza A Virus by Universal Primers and Sequencing by High-throughput Sequencing
Wang Kaicheng1,Qiu Yuan1,Ji Haiwang2,Peng Cheng1,Zhuang Qingye1,Wang Tong1, Wang Suchun1,Yu Jianmin1,Xu Rongqiang1,Chen Jiming1
(1. China Animal Health and Epidemiology Center,Qingdao,Shandong 266032;2. Agricultural Service Center of Wanggezhuang Street in Laoshan District,Qingdao,Shandong 266105)
To sequence the genome of infuenza A virus by high-throughput sequencing,a pair of universal primers was designed to amplify the genome by RT-PCR. The sensibility of the method was also analyzed. The results showed that all the 8 genes of several subtypes infuenza A virus could be amplifed,and the lowest detection limit was 100 pg RNA.
infuenza A virus;universal primers;genome;high-throughput sequencing
book=90,ebook=96
S851.3
A
1005-944X(2017)01-0090-04
10.3969/j.issn.1005-944X.2017.01.025
科技部科技基础性专项(SQ2012FY3260033);中国动物卫生与流行病学中心创新基金(2015IF-0004FF)
陈继明
