中国人群常见恶性肿瘤遗传风险预测模型研究进展
2016-12-09田剑波缪小平林东昕
田剑波缪小平林东昕
(1 华中科技大学同济医学院公共卫生学院流行病与卫生统计学系,武汉 430030;2 中国医学科学院中国协和医科大学肿瘤医院肿瘤研究所病因及癌变研究室,癌发生及预防分子机理北京市重点实验室,北京 100730)
中国人群常见恶性肿瘤遗传风险预测模型研究进展
田剑波1缪小平1林东昕2
(1 华中科技大学同济医学院公共卫生学院流行病与卫生统计学系,武汉 430030;2 中国医学科学院中国协和医科大学肿瘤医院肿瘤研究所病因及癌变研究室,癌发生及预防分子机理北京市重点实验室,北京 100730)

田剑波,硕士,研究方向为流行病与卫生统计。
E-mail:tianjianbo1992@126.com
肿瘤的发生发展是一个涉及多基因、多阶段、多步骤的过程。由于肿瘤表型的高度异质性以及肿瘤病因的复杂性,其风险预测模型构建一直是研究的热点和难点之一。全基因组关联研究(genome wide association studies, GWAS)是分子流行病学研究的重要策略,迄今发现了大量与肿瘤风险相关的易感基因与遗传位点。在传统肿瘤风险预测模型基础上纳入遗传信息,对肿瘤高危人群早期识别、精准预防以及个体化干预具有重大的公共卫生转化意义。文章主要介绍中国人群常见恶性肿瘤遗传风险预测模型的研究现状,以及所面临的机遇和挑战。
WELTER D, MACARTHUR J, MORALES J, et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Research, 2014, 42(Database issue):D1001-1006.
HE J, HU N, ZUO X B, et al. Genomewide association study identifies common variants in SLC39A6 associated with length of survival in esophageal squamous-cell carcinoma. Nature Genetics, 2013, 45(6):632-638.
WEI Q, YU D, LIU M, et al. Genome-wide association study identifies three susceptibility loci for laryngeal squamous cell carcinoma in the Chinese population. Nature Genetics, 2014, 46(10): 1110-1114.
1 中国人群常见恶性肿瘤的全基因组关联研究进展
随着人类基因组计划、人类单倍型计划、千人基因组计划的相继完成,以及全基因组关联研究方法和高通量测序技术的日益成熟,人类已经发现了多个与疾病相关的易感基因和遗传位点。据美国国家人类基因组研究所(National Human Genome Research Institute, NHGRI)统计,目前GWAS已经应用于200余种疾病和400余类性状研究,鉴定了1.5万余个与疾病或性状相关的遗传易1。在肿瘤GWAS研究领域,我国科学家也独立开展了许多具有开创性的工作,并取得了丰硕的原创性成果。中国医学科学院肿瘤研究所林东昕教授课题组在中国人群食管鳞癌、胰腺癌、淋巴瘤和喉癌等常见肿瘤中开展多项GWAS研究,发现多个与肿瘤风险 及预后相关的易感基因和区域感位点7;南京医科大学沈洪兵教授课题组在肺癌、肝癌和胃癌的研究中发现多个易感区域2~10;同时,中国科研工作者还在鼻咽癌、宫颈癌以及前列腺癌等常见肿瘤中开展了GWAS研究,并取得了重要发现8~11~14。
2 中国人群常见恶性肿瘤遗传风险预测模型研究现状
在欧美国家/地区,已联合利用遗传信息和传统环境危险因素在多种疾病中构建了风险预测模型,如冠心病、前列腺癌、膀胱癌,乳腺癌、结直肠癌等,并取得了一定的效果15。中国科研工作者也利用GWAS发现的易感位点在肿瘤学领域开展了一系列的肿瘤风险预测研究,为肿瘤的早期筛选和预防提供了策略和依据(表1)。
2.1 消化系统肿瘤
食管癌是常见的消化道恶性肿瘤之一,发病率居我国恶性肿瘤第3位,死亡率居第4位。食管癌起病隐匿,早期无明显症状,确诊时多数病人已属中晚期,5年生存率仅为15%~25%16。食管鳞癌是具中国特色及地域特征的恶性肿瘤之一,也是目前我国G WAS研究较多的消化系统肿瘤之一。利用GWAS发现的遗传易感位点结合传统的危险因素尝试构建肿瘤风险预测模型,对食管癌的早期预防具有重要意义。笔者课题组在中国人群中利用食管鳞癌GWAS发现的25个易感位点联合年龄、性别、吸烟及饮酒状态4个传统风险因素构建了食管癌风险预测模型,用遗传风险评分(genetic risk score,GRS)的方法计算遗传易感位点联合作用17。结果发现纳入易感位点后预测模型受试者工作特征曲线下面积(评价模型预测能力指标)达到0.709,显著优于仅使用传统风险因素建立的预测模型(AUC=0.639),其AUC水平提高了近7个百分点,显著提高了对食管癌发病风险的预测能力。同时,笔者课题组还利用其中8个与饮酒有交互作用的易感位点单独与传统风险因素联合来预测食管癌发病风险,尽管与传统的预测模型相比,AUC水平仅提高1.2%,但通过加入基因-饮酒的交互作用,依旧可以对模型的灵敏度和特异度有所改善17。此外,笔者课题组又进一步采用了一种新的评估方法,即净再分类改善率来评价纳入GWAS发现的易感位点对食管癌风险预测的作用,发现食管癌风险预测模型由遗传因素、非遗传因素以及基因饮酒交互作用构成的风险预测模型显著优于只包含非遗传因素的预测模型,其净再分类改善率达到了0.086(95% CI: 0.077~0.096)17。
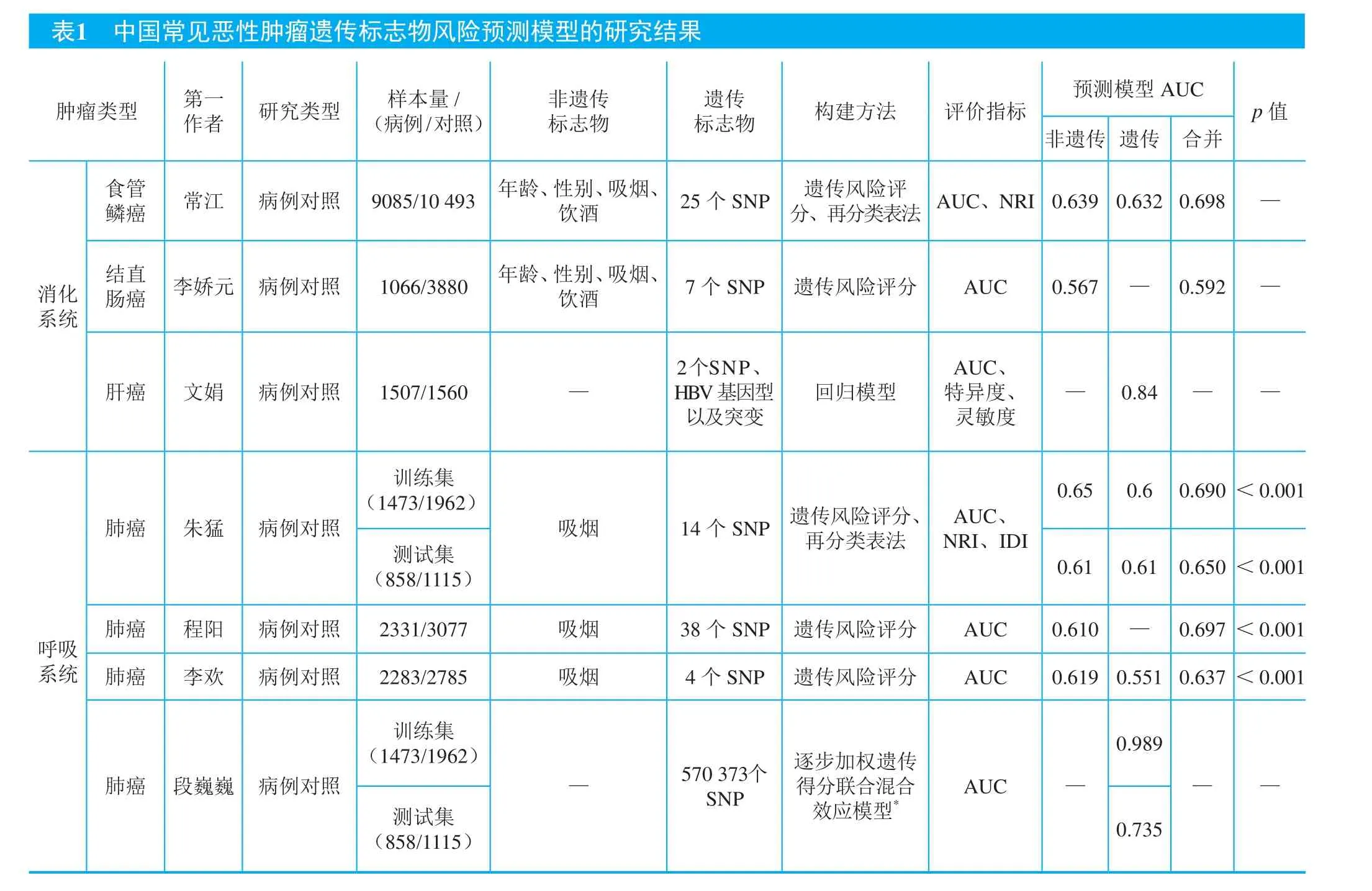
表1 中国常见恶性肿瘤遗传标志物风险预测模型的研究结果肿瘤类型 第一预测模型AUC作者 研究类型 样本量/(病例/对照)非遗传标志物遗传标志物 构建方法 评价指标p值非遗传 遗传 合并食管鳞癌 常江 病例对照 9085/10 493 年龄、性别、吸烟、饮酒 25个SNP 遗传风险评分、再分类表法 AUC、NRI0.6390.632 0.698 —消化系统结直肠癌 李娇元 病例对照 1066/3880 年龄、性别、吸烟、饮酒 7个SNP 遗传风险评分 AUC 0.567 — 0.592 —肝癌 文娟 病例对照 1507/1560 —2个SNP、HBV基因型以及突变回归模型AUC、特异度、灵敏度— 0.84 — —训练集(1473/1962)0.65 0.6 0.690<0.001吸烟 14个SNP 遗传风险评分、再分类表法肺癌 朱猛 病例对照测试集(858/1115)AUC、NRI、IDI 0.61 0.61 0.650<0.001呼吸系统肺癌 程阳 病例对照 2331/3077 吸烟 38个SNP 遗传风险评分 AUC 0.610 — 0.697<0.001肺癌 李欢 病例对照 2283/2785 吸烟 4个SNP 遗传风险评分 AUC 0.6190.551 0.637<0.001训练集(1473/1962)——测试集(858/1115)0.989肺癌 段巍巍 病例对照AUC ——570 373个SNP逐步加权遗传得分联合混合效应模型*0.735

注:AUC(area under the receiver operating characteristic curve,受试者工作特征曲线下面积);SNP(single nucleotide polymorphism,单核苷酸多态性);NRI(net reclassification improvement,净再分类改善率);IDI(integrated discrimination improvement,综合判别优值); AR(absolute risk,绝对危险度);OR(odds ratio,比值比);HBV(hepatitis B virus,乙型肝炎病毒);*代表数据集或者方法之一。
WU C, KRAFT P, ZHAI K, et al. Genomewide association analyses of esophageal squamous cell carcinoma in Chinese identify multiple susceptibility loci and gene-environment interactions. Nature Genetics, 2012, 44(10):1090-1097.
WU C, MIAO X, HUANG L, et al. Genomewide association study identifies five loci associated with susceptibility to pancreatic cancer in Chinese populations. Nature Genetics, 2012, 44(1):62-66.
WU C, WANG Z, SONG X, et al. Joint analysis of three genome-wide association studies of esophageal squamous cell carcinoma in Chinese populations. Nat Genet, 2014, 46(9):1001-1006.
WU C, XU B, YUAN P, et al. Genome-wide interrogation identifies YAP1 variants associated with survival of small-cell lung cancer patients. Cancer Research, 2010, 70(23):9721-9729.
JIN G, MA H, WU C, et al. Genetic variants at 6p21.1 and 7p15.3 are associated with risk of multiple cancers in Han Chinese. American Journal of Human Genetics, 2012, 91(5):928-934.
在其他消化道肿瘤,如结直肠癌、肝癌等,我国科学家也尝试利用遗传易感位点建立相应的肿瘤风险预测模型。笔者课题组在中国人群中运用7个与中国人群结直肠癌相关的易感位点联合年龄、性别、吸烟、饮酒等传统危险因素构建结直肠癌风险预测模型,结果发现纳入易感位点可显著提高模型对结直肠癌的风险预测能力(AUC=0.592),优于由单一的传统风险因素构建的预测模型(AUC=0.567),提示在肿瘤风险预测模型中纳入遗传标志物对结直肠癌发病风险评估有较大的改善18。此外,南京医科大学胡志斌教授课题组利用HLA区域上的遗传变异(rs9272105、rs92753)慢性乙型肝炎病毒的基因型及突变对肝细胞癌发病进行风险评估,达到较好的预测效果(AUC=0.840)19。
2.2 呼吸系统肿瘤
肺癌是我国发病率和死亡率最高的恶性肿瘤之一,由于早期诊断率低,5年生存率不到10%。早期发现与早期治疗是提高肺癌诊治效果、降低死亡率、提高生存率的有效途径。构建肺癌发病风险预测模型,实现肺癌的早期预防和干预成为一个亟待解决的医学挑战。自2008年以来,国内外已开展了多项肺癌GWAS研究21,鉴别出了20多个易感区域和40多个易感位点,为肺癌遗传风险预测模型的构建提供了有利条件2023。在我国,南京医科大学沈洪兵教授课题组利用两个独立的研究人群分别作为训练集和验证集,前者进行参数的估计和模型的构建,后者对模型进行评估,结果表明联合14个易感位点可以将吸烟预测肺癌风险的效能(评价指标为AUC)从0.60提高到0.65,同时另两个评价风险预测能力的指标,净分类指数和整体鉴别指数也有一定程度的提高,较单纯依据吸烟量因素显著提高肺癌预测能力2224。李欢、程阳等也分别在中国人群中利用遗传风险评分的方法评价遗传易感位点对肺癌发病风险的预测能力,结果发现,相比传统肺癌风险预测模型,纳入易感位点均可不同程度提高模型的AUC值,提高模型的预测效能26。此外,针对因GWAS易感位点筛选统计检验标准过严,导致一些低效应易感位点未被充分挖掘,南京医科大学陈峰教授课题组对肺癌易感位点进行深度处理,形成一个多阶段的风险预测分析策略,在两个独立人群中25比较了6种新的模型构建方法,其中发现逐步加权遗传得分(stepwise weighted GRS, sGRS)和混合效应模型(linear mixed models, LMMs)效果最为明显,sGRS-LMMs联合的方法可将肺癌风险预测模型曲线下面积在训练集提高到0.989,在验证集中也达到了0.735,大幅度提高了肺癌的预测能力和准确度27。这几项研究都提示联合遗传易感位点可在不同程度上提高、改善肺癌风险预测模型的预测效能。
2.3 其他常见恶性肿瘤
SHI Y, HU Z, WU C, et al. A genome-wide association study identifies new susceptibility loci for non-cardia gastric cancer at 3q13.31 and 5p13.1. Nature Genetics, 2011, 43(12):1215-1218.
10 参考文献
HU Z, WU C, SHI Y, et al. A genome-wide association study identifies two new lung cancer susceptibility loci at 13q12.12 and 22q12.2 in Han Chinese. Nature Genetics, 2011, 43(8):792-796.
11 参考文献
ZHANG H, ZHAI Y, HU Z, et al. Genomewide association study identifies 1p36.22 as a new susceptibility locus for hepatocellular carcinoma in chronic hepatitis B virus carriers. Nature Genetics, 2010, 42(9):755-758.
12 参考文献
BEI J X, LI Y, JIA W H, et al. A genome-wide association study of nasopharyngeal carcinoma identifies three new susceptibility loci. Nature Genetics, 2010, 42(7):599-603.
13 参考文献
SHI Y, LI L, HU Z, et al. A genome-wide association study identifies two new cervical cancer susceptibility loci at 4q12 and 17q12. Nature Genetics, 2013, 45(8):918-922.
14 参考文献
XU J, MO Z, YE D, et al. Genome-wide association study in Chinese men identifies two new prostate cancer risk loci at 9q31.2 and 19q13.4. Nature Genetics, 2012, 44(11):1231-1235.
15 参考文献
CHATTERJEE N, SHI J, GARCIA-CLOSAS M. Developing and evaluating polygenic risk prediction models for stratified disease prevention. Nature Reviews Genetics, 2016, 17(7):392-406.
16 参考文献
CHEN W, ZHENG R, BAADE P D, et al. Cancer statistics in China, 2015. CA: A Cancer Journal for Clinicians, 2016, 66(2):115-132.
17 参考文献
CHANG J, HUANG Y, WEI L, et al. Risk prediction of esophageal squamous-cell carcinoma with common genetic variants and lifestyle factors in Chinese population. Carcinogenesis, 2013, 34(8):1782-1786.
18 参考文献
李娇元, 缪小平, 林东昕. 中国常见肿瘤的全基因组关联研究进展. 自然杂志, 2015, 37(1):1-7.
中国科学家还在其他恶性肿瘤中尝试应用GWAS发现的易感位点构建遗传风险预测模型。乳腺癌是女性最常见的恶性肿瘤之一,其发病率呈快速增长趋势。Zheng等28在中国人群中利用易感位点预测乳腺癌的发病风险,该研究包括3039例乳腺癌病例和3082例对照,结果显示,单纯利用初潮年龄、首胎活产年龄、腰臀比、乳腺癌肿瘤家族史和乳腺良性疾病史等传统因素构建模型,其对乳腺癌发病风险的预测能力为0.618(AUC),在增加了8个遗传易感位点的信息后,预测能力提高至0.630(AUC)。戴俊程等29利用与中国人群乳腺癌风险相关的5个GWAS易感位点,联合乳腺癌传统危险因素(初潮年龄、首次活产年龄)构建并评价风险预测模型,结果同样发现在模型中纳入易感位点能够改善传统风险预测模型30介绍了在我国人群中进行乳腺癌遗传风险评价的研究思路、实施流程以及研究现状。同时,复旦大学徐剑锋教授课题组的预测能力。黄育北等31在膀胱癌中构建了遗传风险预测模型,结果发现纳入GWAS易感位点能显著改善模型对膀胱癌的预测能力。总体而言,目前利用易感位点来辅助传统危险因素预测乳腺癌或者膀胱癌等恶性肿瘤发病风险,尽管有一定的提升,但效果不明显,还有很大的上升空间。
19 参考文献
WEN J, SONG C, JIANG D, et al. Hepatitis B virus genotype, mutations, human leukocyte antigen polymorphisms and their interactions in hepatocellular carcinoma: a multi-centre casecontrol study. Scientific Reports, 2015, 5:16489.
20 参考文献
SHIRAISHI K, KUNITOH H, DAIGO Y, et al. A genome-wide association study identifies two new susceptibility loci for lung adenocarcinoma in the Japanese population. Nature Genetics, 2012, 44(8):900-903.
21 参考文献
DONG J, HU Z, WU C, et al. Association analyses identify multiple new lung cancer susceptibility loci and their interactions with smoking in the Chinese population. Nature Genetics, 2012, 44(8):895-899.
22 参考文献
WEISSFELD J L, LIN Y, LIN H M, et al. Lung cancer risk prediction using common SNPs located in GWAS dentified susceptibility regions. 2015, 10(11):1538-1545.
23 参考文献
HOGGART C, BRENNAN P, TJONNELAND A, et al. A risk model for lung cancer incidence. Cancer Prev Res (Phila), 2012, 5(6):834-846.
24 参考文献
ZHU M, CHENG Y, DAI J, et al. Genomewide association study based risk prediction model in predicting lung cancer risk in Chinese. Zhonghua Liu Xing Bing Xue Za Zhi, 2015, 36(10):1047-1052.
25 参考文献
LI H, YANG L, ZHAO X, et al. Prediction of lung cancer risk in a Chinese population using a multifactorial genetic model. BMC Medical Genetics, 2012, 13:118.
26 参考文献
CHENG Y, JIANG T, ZHU M, et al. Risk assessment models for genetic risk predictors of lung cancer using two-stage replication for Asian and European populations. Oncotarget, 2016.
27 参考文献
段巍巍, 赵杨, 张丽伟, 等. 使用肺癌GWAS数据进行遗传风险预测的方法和策略研究. 中国卫生统计, 2015, 32(4):554-557.
28 参考文献
ZHENG W, WEN W, GAO Y T, et al. Genetic and clinical predictors for breast cancer risk assessment and stratification among Chinese women. Journal of the National Cancer Institute, 2010, 102(13):972-981.
此外,上海复旦大学卢大儒教授课题组32利用与中国人群神经胶质瘤相关的GWAS易感位点,联合家族史等传统危险因素构建了胶质瘤风险预测模型。采用了3种不同的方法,即计数遗传风险评分、权重遗传风险评分以及logistic回归风险评分来计算易感位点的联合作用,结果发现利用logistic回归风险评分方法联合易感位点和胶质瘤家族史构建的预测模型对胶质瘤的风险预测能力最强,AUC值达到0.646,研究结果对神经胶质瘤患者的早期识别具有一定的意义。
3 总结 和展望
综合现有的研究成果,在原有传统风险预测模型的基础上加入遗传易感因素,确实可以提高某些肿瘤风险模型的预测能力,但提高的幅度仍十分有限。这一方面是由于肿瘤的遗传易感因素尚未彻底阐明,纳入的疾病易感位点不够系统全面,另一方面也与模型的构建、评价方法不够优化、系统有关。此外,人群的异质性,以及目前的模型构建主要是基于病例对照研究,这些都可能在一定程度上存在流行病学偏倚,从而造成遗传预测效果的不可重复性和失真。同时,目前的遗传风险预测模型尚未充分考虑基因-基因以及基因-环境之间的交互作用。我国常见恶性肿瘤遗传风险预测模型的研究仍存在以下机遇与挑战。
3.1 遗传易感位点的系统深度挖掘
GWAS是基于常见疾病常见变异(common disease-common variant, CDCV)假设的研究方法,所涉及的遗传变异的MAF一般大于5%,尚未充分发现一些与疾病相关的低频变异或者罕见变异,这可能会造成疾病的遗传度部分丢失33。对全基因组或者全外显子组深度测序以及对某些有潜在功能的特定区域的重测序和精细定位分析可能是GWAS下一阶段的重要任务3435。此外,目前GWAS研究策略通常选用较为严格的标准(p≤10-7)去筛选出具有统计学差异的SNP进行第二阶段的验证。这在降低第一阶段筛选假阳性的同时,也可能会丢失一批遗传易感位点。适当放宽SNP的筛选标准,扩大验证范围,可能会发现更多疾病易感位点。
3.2 基因-环境交互作用
肿瘤等复杂疾病大多不是单纯由遗传因素或者环境因素决定的,基因-基因以及基因-环境之间的交互作用在肿瘤发生、发展中起着重要作用。笔者课题组曾结合饮酒数据在中国汉族人群中进行了食管鳞状细胞癌的全基因组基因-环境交互效应分析,结果发现相比于不携带变异基因型的饮酒者,同时携带ADH1B和ALDH2基因特定位点变异的饮酒者,其罹患食管鳞癌的风险增加了4倍5。因此,对交互作用的研究探讨有助于认识复杂疾病或性状的遗传机制,对建立有效的风险预测模型有着重大的意义。但目前在大部分肿瘤风险预测模型中并没有考虑基因-基因和基因-环境之间的交互作用,这部分效应的缺失在很大程度上会影响模型的预测效能和准确性,所以在构建风险预测模型时这是尤其需要注意以及提高的一个方向。
3.3 前瞻性队列人群验证
目前肿瘤风险预测模型大部分基于病例对照研究。根据疾病结局来构建的模型并不是严格意义上的预测模型,同时病例对照研究有一定的局限性,如危险因素和疾病结局无法确定因果,选择研究对象时可能存在选择偏倚,在疾病影响因素的收集过程可能存在回忆偏倚等。这些局限均可能在一定程度上影响预测模型的准确性和可靠性。因此,在大规模多中心的前瞻性队列研究中对风险预测模型进行验证,或者直接在队列研究中构建风险预测模型才能更准确地获知模型的实际预测效果,也是今后的研究方向。
虽然目前肿瘤遗传风险预测模型发展还不够成熟,但其也成功应用于对某些肿瘤高危人群的筛选,能够在发病前对不同风险人群采取差异化的干预策略。如Kader等36在对前列腺癌的癌前筛选时,依据遗传风险评分把对象划分为不同的风险等级,确定是否需要前列腺穿刺。结果发现依据风险评分来判断是否需要前列腺穿刺与单纯依据传统临床因素相比,降低了15%的过度穿刺,避免了医疗资源的浪费,这在一定程度上反映了遗传风险预测模型在肿瘤防治领域的转化医学意义。
总之,以中国人群为对象的常见恶性肿瘤GWAS研究取得了丰硕的成果,不仅有利于更全面地解读肿瘤遗传病因和致病机制,同时也为构建更有效的肿瘤遗传风险预测模型提供了新的线索和依据。但是,中国人群的肿瘤遗传易感因素尚未彻底阐明,基因-环境交互作用有待进一步挖掘,构建具有转化意义的常见恶性肿瘤遗传风险预测模型仍需要中国科学家持续不断的努力和创新。
[本研究获得国家自然科学基金(81222038)、霍英东青年教师基金(131038)、国家重点研发计划(2016YFC1302702)资助。]
29 参考文献
DAI J, HU Z, JIANG Y, et al. Breast cancer risk assessment with five independent genetic variants and two risk factors in Chinese women. Breast Cancer Research, 2012, 14(1):R17.
30 参考文献
HUANG Y, SONG F, CHEN K. Current status of genome-wide association studies (GWAS) on breast cancer and application values of single nucleotide polymorphisms identified from GWAS. Zhonghua Liu Xing Bing Xue Za Zhi, 2015, 36(10):1058-1061.
31 参考文献
王萍. 膀胱癌遗传分数的计算及发病风险预测模型的构建. 上海: 复旦大学, 2013. www. cnki.net/KCMS/detail/detail.aspx?QueryID=0&Cu rRec=2&recid=&filename=1015419704.nah&dbn ame=CMFD201601&dbcode=CMFD&pr=&urlid= &yx=&v=MDk5MDhGOWJNcTVFYlBJUjhlWDF MdXhZUzdEaDFUM3FUcldNMUZyQ1VSTHllW nVSckZ5L25VNy9NVkYyNkc3ZTU=.
32 参考文献
ZHAO Y, CHEN G, YU H, et al. Development of risk prediction models for glioma based on genome-wide association study findings and comprehensive evaluation of predictive performances. Oncotarget, 2016.
33 参考文献
SHI J, YANG X R, BALLEW B, et al. Rare missense variants in POT1 predispose to familial cutaneous malignant melanoma. Nature Genetics, 2014, 46(5):482-486.
34 参考文献
PARK H, KIM J I, JU Y S, et al. Discovery of common Asian copy number variants using integrated high-resolution array CGH and massively parallel DNA sequencing. Nature Genetics, 2010, 42(5):400-405.
35 参考文献
CIRULLI E T, GOLDSTEIN D B. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nature Reviews Genetics, 2010, 11(6):415-425.
36 参考文献
KADER A K, SUN J, RECK B H, et al. Potential impact of adding genetic markers to clinical parameters in predicting prostate biopsy outcomes in men following an initial negative biopsy: findings from the REDUCE trial. European Urology, 2012, 62(6):953-961.
10.3969/j.issn.1674-0319.2016.06.002
