高免疫球蛋白D伴周期性发热综合征1例病例报告
2016-11-26刘丹如朱晓华王宏胜孙金峤翟晓文王晓川
侯 佳 刘丹如 朱晓华 王宏胜 孙金峤 翟晓文 王晓川
·论著·
高免疫球蛋白D伴周期性发热综合征1例病例报告
侯 佳1刘丹如1朱晓华2王宏胜2孙金峤1翟晓文2王晓川1
目的 提高对高IgD伴周期性发热综合征(HIDS)的认识。方法 回顾性总结1例儿童HIDS病例的临床特征、实验室检查、血清IgD、MVK酶和MVK基因检测结果。结果 6岁7个月男孩,2岁起病,呈周期性发作性发热,每2~4周发热1次,每次持续3~7 d。发热时伴腹痛,腹泻,关节疼痛,肝脾肿大。发热时未用抗生素经退热对症治疗,体温可恢复正常。发热时WBC、N和CRP升高,热退后可降至正常。免疫接种后有发热和感染史。经全面检查排除感染性、风湿性及血液肿瘤相关疾病。MVK基因分析发现外显子11,c.1129G>A,p.V377I,为杂合型错义突变,外显子9,c.790_791insC,p.Leu264fsX12,为杂合型插入突变(首次报道的新突变)。患儿血清IgD(1 084 μg·mL-1)显著高于正常参照值(<100 μg·mL-1)。MVK酶(23 ng·mL-1)低于正常参照值(50~300ng·mL-1)。本文病例临床特征典型,HIDS诊断明确。结论 婴儿期起病的周期性发作性发热,需警惕HIDS可能,免疫接种后发热是重要的诊断线索,检测血清IgD和MVK酶水平是重要诊断依据,MVK基因突变可明确诊断。
高IgD综合征; 周期性发热; 甲羟戊酸激酶缺陷; 自身炎症性疾病
1 病例资料
男,6岁7月,汉族。因“间歇发热4年”入复旦大学附属儿科医院(我院)。患儿系孕7月引产儿,出生体重1.8 kg,无窒息缺氧史,生后4 d因发热于当地ICU治疗1周后好转出院。被现任父母抱养,故家族史不详;家长述疫苗接种后有发热,接种百白破疫苗后有1次化脓感染史。
患儿自2岁开始出现间断发热,每月1~2次,每次发热持续6~7 d,体温最高40℃,血WBC升高,发热前有寒战,有时伴咳嗽、流涕、腹痛和腹泻,无皮肤黄染、皮疹、头痛、呕吐、抽搐和关节肿痛。在当地医院抗感染、退热治疗后可好转。3岁5个月因“发热”曾住他院治疗,血WBC 8.6×109·L-1,N 0. 45,Hb 125 g·L-1,PLT 386×109·L-1,CRP 11 mg·L-1;超声心动图示左室内径轻度增大,心电图示左心室高电压、V4~V6导联ST段下移,心肌酶谱正常;单纯疱疹病毒IgM(+),抗核抗体和抗双链DNA抗体(-);骨髓细胞形态学:骨髓增生活跃,粒系各阶段比值大致正常,成熟阶段细胞可见中毒颗粒,红系增生尚可,粒红比例偏低,部分中晚幼可见胞浆少,嗜碱性强,成熟RBC大小不等,以小为主,轻度中空,有变形,巨核细胞和PLT不减少。诊断感染性心肌炎,予抗感染、营养心肌治疗后,复查心电图和超声心动图正常。出院后患儿仍有间断发热,频率同前。入住我院前2 d,患儿再次发热,体温最高40℃,口服退热药后可缓解,伴有腹痛和腹泻,门诊拟“发热待查”收住我院。
入院查体:神清,呼吸平稳,精神反应可,体型匀称;未见皮疹及出血点,颌下可触及数枚0.5 cm×0.5 cm淋巴结,质软,活动度可,无触痛,咽部未见充血,双侧扁桃体未见肿大,甲状腺未扪及,颈软;双肺呼吸音清,未闻及干湿啰音,心率90次/分,心音有力律齐,未闻及病理性杂音;腹软,肝肋下3 cm,脾肋下1.5 cm,质软,无触痛,未及异常包块;四肢肌力肌张力正常,关节无红肿热痛,神经系统检查未见异常。
一般实验室检查:发热时WBC 13.4~21.1×109·L-1,N 0.78~0.86,CRP 83 mg·L-1;体温正常时WBC 4.5~8.5×109·L-1,N 0.36~0.44,CRP<8 mg·L-1。外周血涂片未见异常淋巴细胞,血沉20 mm·h-1,降钙素原0.56 ng·mL-1,Hb、PLT、内毒素、尿粪常规、肝肾功能、电解质、心肌酶谱、凝血功能、抗O、类风湿因子、自身免疫抗体和微量元素检查均正常。
影像学检查:腹部B超检查显示肝脾肿大(肝肋下3.0 cm、剑突下 5.0 cm、脾肋下1.5 cm),腹腔内淋巴结轻度肿大,胰腺、双肾、双侧肾上腺区和后腹膜未见局灶性占位。心电图、心脏超声、头颅MRI增强、胸腹和盆腔CT增强均未发现异常。
病原学检测:血培养、尿培养、骨髓细菌培养、结核抗体、T-spot阴性、TORCH、EBV抗体、EBV-DNA、CMV-DNA、单纯疱疹病毒Ⅰ-DNA、单纯疱疹病毒Ⅱ-DNA、肺炎支原体IgM、真菌葡聚糖、肥达试验和寄生虫抗体检测均阴性;乙肝二对半、丙肝抗体、梅毒和HIV抗体检测未见异常。
免疫功能评估: IgG 6.64(4.95~12.74)g·L-1,IgA 2.86(0.33~1.89)g·L-1,IgM 0.49(0.65~2.01)g·L-1,总IgE 89.2(<100)KUA·L-1; CD3+83.5(64~73)%,CD3+CD8+25.5(24~34)%,CD3+CD4+48.5(29~36)%,CD16+CD56+4.4(11~23)%,CD19+11.1(14~21)%。补体和中性粒细胞呼吸爆发功能均正常。
遗传代谢和血液肿瘤检测:血串联质谱和尿气相质谱未见异常;骨髓穿刺示骨髓增生极度活跃,粒红比减低,粒系有毒性变。
患儿入院前后呈现较有规律的发热现象(图1),2周左右1次发热、持续3 d左右,体温39℃左右;入院后未使用抗生素,仅予降温对症处理,患儿体温可自行恢复正常;体温升高与血WBC、N比例和CRP升高密切相关;发热时伴腹痛、腹泻、关节痛和肝脾肿大;疫苗接种后有发热和感染病史;排除了感染性、风湿性和血液肿瘤性疾病。因此怀疑自身炎症性疾病高IgG伴周期性发热综合征(HIDS),行甲羟戊酸激酶(MVK)基因检测。
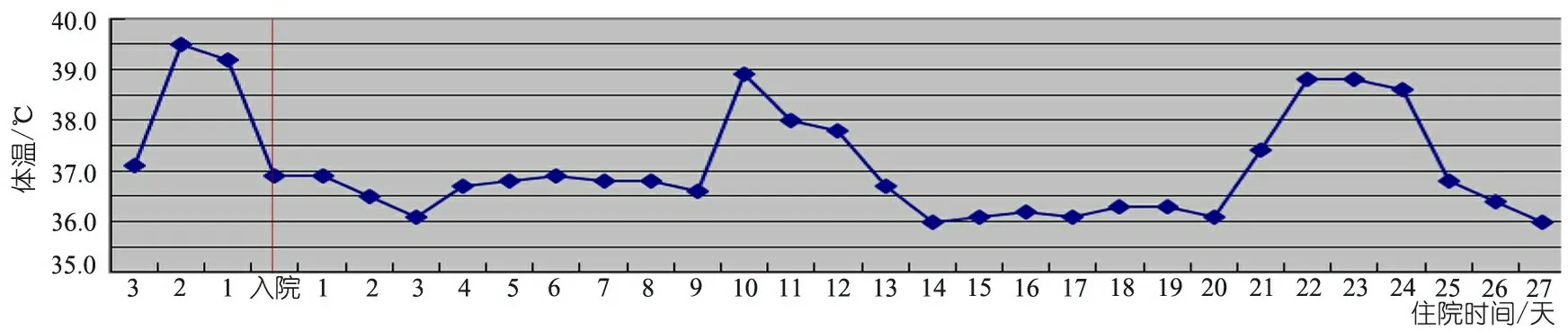
图1 患儿住院期间体温变化
标准方法提取外周血DNA,PCR扩增MVK基因10个编码外显子区及邻近序列,引物见表1,PCR反应条件:95℃ 5min,随后95℃ 30 s、60℃ 30 s、72℃ 40 s,共35个循环,72℃ 7 min。产物纯化后直接测序。所有测序结果进行机器及人工读图,并与基因库中标准序列进行比较。患儿MVK基因有2个突变位点,第11外显子,c.1129G>A,p.V377I,杂合型错义突变;第9外显子,c.790_791insC,p.Leu264fsX12,杂合型插入突变导致移码,为首次报道的新突变位点(图2)。图3显示,2个突变位点在MVK蛋白模型上的三维空间位置。
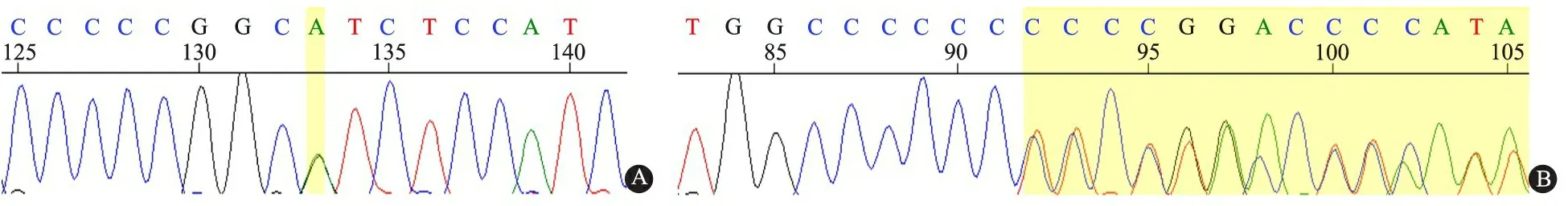
图2 本例患儿MVK基因分析结果
注 A:第11外显子,c.1129G>A,p.V377I,杂合型错义突变(黄色区域位点);B:第9外显子,c.790_791insC,p.Leu264fsX12,杂合型插入突变导致移码(黄色区域位点)

表1 MVK基因检测引物序列
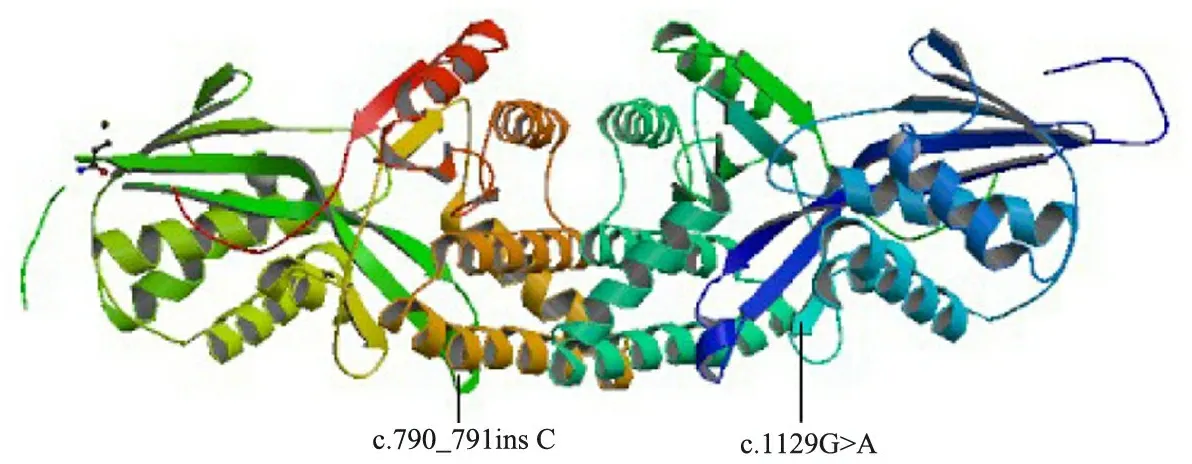
图3 两个突变位点在MVK蛋白模型三维空间的位置
本文发热待查病例HIDS诊断基本成立,为进一步从IgD表达和MVK酶表达水平验证HIDS诊断,行血清IgD(ELISA法,Human ELISA Kit,英国Abcam公司)和MVK酶(ELISA法检测,MVK Human ELISA Kit,武汉华美生物工程有限公司)表达检测。基于同期、同一实验室、同一检测方法对5~7岁72名正常儿童建立参照值,本文患儿血清IgD 1 084 μg·mL-1明显高于正常儿童参照值(<100 μg·mL-1),MVK 23 ng·mL-1低于正常儿童参照值(50~300 ng·mL-1)。
2 讨论
HIDS(OMIM 260920)是一种罕见的自身炎症性疾病,呈常染色体隐性遗传。至今全世界有数百例报道,大部分是白人,约60%为荷兰裔和法国裔[1],国内尚无基因确诊的病例报道。HIDS易感基因外显率较低(1∶350),患儿双亲或后代常无此病。存在无临床症状而未被诊断的病例,HIDS发病率可能被低估。HIDS的诊断年龄有显著的滞后,平均为7.1年[2],与对本病的认识不足和较复杂的排他性诊断有关。
基于国外病例系列报告10例以上的文献复习[3~7],HIDS常在1岁内发病,平均起病年龄为4~10月龄。临床特征为周期性发热,几乎所有患儿都有发作性发热症状,每次发热持续时间1~10 d,典型者每次发作3~7 d, 4~6周发作1次,个体差异大。42%~73%患儿有发作诱因,感染和疫苗接种可以诱发,也有报道应激为诱发因素。高热前常有寒战,发热时常伴有的症状(发生频率>50%)包括:腹痛、呕吐、腹泻、皮疹、关节痛、肌痛、头痛及口疮性口炎。发作时可触及肿大淋巴结,颈部最多见,在年轻患者中有脾肿大。许多患儿有非破坏性的反复关节炎,主要累及大关节[3~7]。部分患儿有色素性视网膜炎[8,9]。文献报道有少数患儿发生淀粉样变性(4%),常发生在发病18年后[3]。发作间隔期患儿可完全健康,约71%患儿有乏力不适全身症状。约25%患者有情绪异常,提示该疾病存在一定心理影响[6]。随年龄增长,发热发作频率和程度趋于减少或降低。发作时有急性期反应物升高,包括WBC和N增多,ESR升高。血清IgD水平升高(>100 U·mL-1),大部分患儿也有血清IgA升高。各种免疫接种引发的反复发作性发热是早期诊断的重要线索。本例患儿周期性发热特征明显,未使用抗生素,仅予降温对症处理下患儿体温可自行恢复正常;排除导致发热性疾病相关检查充分且依据较强,存在免疫接种后发热和感染的病史,符合HIDS临床特点。
婴儿期周期性发热如果临床症状提示上述表现,应怀疑HIDS并行血清IgD和IgA检测,如果2个都升高,可临床初步诊断。但血清IgD升高并非持续存在,特别是<3岁的婴幼儿。有文献报道12%~28%的HIDS患儿IgD不升高[5,10]。本例患儿血清IgA 和IgD 显著高于同年龄段正常值。尿液甲羟戊酸检测是一种敏感的筛查MKD方法,约93%的患儿排泄的甲羟戊酸量升高[5,11]。尽管国外有报道在明确MKV基因致病突变并且尿甲羟戊酸含量增高的患儿中发现甲羟戊酸酶水平无异常,但多数文献仍然认为甲羟戊酸酶测定是诊断MKD的金标准。本例患儿采用ELISA方法检测MVK酶水平低下,HIDS诊断明确。
在发现MVK为致病基因之前,血清IgD升高是临床诊断HIDS所必需选项,但有数例MVK基因突变而血清IgD水平正常的HIDS病例报道,因此筛查MVK基因突变具有确诊价值。目前认为HIDS是由MVK基因突变所致。MVK基因定位于12q24,包含10个编码外显子和1个非编码外显子,约21 kb,编码甲羟戊酸激酶[12]。MVK是胆固醇合成的关键酶之一,使甲羟戊酸磷酸化生成磷酸甲羟戊酸,后者进一步被催化合成类异戊二烯和胆固醇。MVK基因突变可影响
其酶的活性及稳定性,使甲羟戊酸激酶活性降到正常的5%~15%,若MVK活性完全丧失则引起甲羟戊酸尿症[13]。MVK基因突变导致炎症反应的机制仍不清楚,推测甲羟戊酸堆积及类异戊二烯减少导致IL-1β分泌异常增高,从而引起炎症反应[14,15]。
对MVK突变基因型分析发现,最常见的突变位点为V377I,其次为I268T[3,4,16]。最常见的突变类型为p.V377I/I268T复合杂合突变,约占22%,其次为p.V377I純合突变,约占11%。而p.V377I/p.I268T复合杂合突变患儿临床表现往往较重,更易出现淀粉样变性病变。本文报道患儿MVK基因检测发现2个突变位点,系复合杂合突变,其中有已报道的最常见的V377I,另一个位点为位于第9外显子的杂合型插入突变,c.790_791insC,p.Leu264fsX12,导致移码,为首次报道的新突变位点。遗憾的是患儿系抱养儿,无法找寻到亲生父母,未能检测亲生父母的MVK基因,因此患儿2个突变位点系自发突变抑或遗传自父母无法确定。
尽管认为HIDS是一种良性疾病,大多为对症治疗。抗炎药物对抑制发热发作效果不一,类固醇对控制病情反复发作无效,而非甾类抗炎药仅部分有效,秋水仙碱可以延长发作间期,但不能减轻病情的严重程度。已有报道,辛伐他汀(HMG-CoA还原酶抑制剂)、阿那白滞素(anakinra,白介素-1受体拮抗剂)以及依那西普(肿瘤坏死因子α抑制剂)对HIDS治疗有一定效果[17]。本例患儿在发热发作时,给予布洛芬治疗后好转,偶有头痛、关节痛表现,随访2年一般状况良好,可以正常上学生活。
[1]Drenth JP, van der Meer JW. Hereditary periodic fever. N Engl J Med,2001,345(24):1748-1757
[2]Berody S, Galeotti C, Koné-Paut I, Piram M. A restrospective survey of patients's journey before the diagnosis of mevalonate kinase deficiency. Joint Bone Spine, 2015, 82(4):240-244
[3]Ter Haar NM, Jeyaratnam J, Lachmann HJ, et al. The phenotype and genotype of mevalonate kinase deficiency: A series of 114 cases from the Eurofever Registry. Arthritis Rheumatol. 2016 May 23. doi: 10.1002/art.39763
[4]Durel CA, Aouba A, Bienvenu B, et al. Observational study of a French and Belgian multicenter cohort of 23 patients diagnosed in adulthood with mevalonate kinase deficiency. Medicine (Baltimore), 2016, 95(11):e3027
[5]Bader-Meunier B, Florkin B, Sibilia J, et al. Mevalonatekinase deficiency: a survey of 50 patients. Pediatrics, 2011, 128(1):e152-159
[6]van der Hilst JC, Bodar EJ, Barron KS, et al. Long-term follow-up, clinical features, and quality of life in a series of 103 patients with hyperimmunoglobulinemia D syndrome. Medicine (Baltimore),2008,87(6):301-310
[7]D'Osualdo A, Picco P, Caroli F, et al. MVK mutations and associated clinical features in Italian patients affected with autoinflammatory disorders and recurrent fever. Eur J Hum Genet, 2005, 13(3):314-320
[8]Balgobind B, Wittebol-Post D, Frenkel J. Retinitis pigmentosa in mevalonate kinase deficiency. J Inherit Metab Dis,2005,28(6):1143-1145
[9]Siemiatkowska AM, van den Born LI, van Hagen PM, et al. Mutations in the Mevalonate Kinase (MVK) Gene Cause Nonsyndromic Retinitis Pigmentosa. Ophthalmology,2013,120(12):2697-2705
[10]Ammouri W, Cuisset L, Rouaghe S, et al. Diagnostic value of serum immunoglobulinaemia D level in patients with a clinical suspicion of hyper IgD syndrome. Rheumatology (Oxford), 2007, 46(10):1597-1600
[11]Jeyaratnam J, Ter Haar NM, de Sain-van der Velden MG, et al. Diagnostic value of urinary mevalonic acid excretion in patients with a clinical suspicion of Mevalonate Kinase Deficiency (MKD). JIMD Rep, 2016,27:33-38
[12]Drenth JP, Cuisset L, Grateau G, et al. Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. International Hyper-IgD Study Group. Nat Genet,1999,22(2):178-181
[13]Haas D, Hoffmann GF. Mevalonate kinase deficiencies: from mevalonic aciduria to hyperimmunoglobulinemia D syndrome. Orphanet J Rare Dis,2006,1:13
[14]Kanazawa N, Furukawa F. Autoinflammatory syndromes with a dermatological perspective. J Dermatol,2007,34(9):601-618
[15]Long SS. Distinguishing among prolonged, recurrent, and periodic fever syndromes: approach of a pediatric infectious diseases subspecialist. Pediatr Clin North Am,2005,52(3):811-835
[16]Cuisset L, Drenth JP, Simon A, et al. Molecular analysis of MVK mutations and enzymatic activity in hyper-IgD and periodic fever syndrome. Eur J Hum Genet,2001,9(4):260-266
[17]Topaloglu R, Ayaz NA, Waterham HR, et al. Hyperimmunoglobulinemia D and periodic fever syndrome; treatment with etanercept and follow-up. Clin Rheumatol,2008,27(10):1317-1320
(本文编辑:张崇凡)
Hyper-IgD and periodic fever syndrome: a case report and literature review
HOU Jia1, LIU Dan-ru1, ZHU Xiao-hua2, WANG Hong-sheng2,SUN Jin-qiao1, ZHAI Xiao-wen2, WANG Xiao-chuan1
(Department of Clinical Immunology, Children's Hospital of Fudan University, Shanghai 201102, China)
WANG Xiao-chuan,E-mail: xchwang@shmu.edu.cn; ZHAI Xiao-wen,E-mail: zhaixiaowendy@163.com
Objective To improve the recognition of hyper-IgD and periodic fever syndrome (HIDS).MethodsThe clinical features, laboratory examinations , serum IgD and MVK level, and alsoMVKgene detection results were retrospectively summarized in HIDS patient. ResultsA 6.6 years old boy was admitted to our hospital due to periodic fever from 2 years old. The periodic episodes of fever occurred every 2 to 4 weeks, and lasted for 3 to 7 days everytime, accompanied with abdominal pain, diarrhea, joint pain and with liver and spleen enlargement. The fever could spontaneously resolved without antibiotic treatment. The amount of WBC, neutrophil and CRP increased with the fever, and decrease to the normal level once the fever resolved. The patients had the history of fever episodes and infection symptom after immunization. The infectious disease, rheumatic disease and neoplastic hematologic disorder were ruled out by detailed and systemic examination. The pathogenic compound heterozygous mutations ofMVKgene:,p.V377I (c.1129G>A) and p.Leu264fsX12 (c.790_791insC, novel mutation) were found and HIDS was diagnosed. The much higher serum level of IgD (1 084 μg·mL-1) and lower MVK level (23 ng·mL-1) were further detected and HIDS was confirmed.ConclusionThe clinical diagnosis of HIDS should be suspected in cases with periodic fever onset from infant period. Immunization induced fever episode was the important clue of the HIDS diagnosis. The levels of serum IgD and MVK were the diagnostic basis and theMVKgene mutation could clarify the HIDS diagnosis.
Hyper IgD syndrome; Periodic fever; Mevalonate kinase deficiency; Hereditary autoinflammatory disease
复旦大学附属儿科医院 1 临床免疫科;2 血液肿瘤科 上海,201102
王晓川,E-mail:xchwang@shmu.edu.cn;翟晓文,E-mail:zhaixiaowendy@163.com
10.3969/j.issn.1673-5501.2016.05.012
2016-08-30
2016-10-18)
