一种新型肿瘤组织起源分子标志物的建立与评价
2016-11-17王奇峰徐清华陈金影钱琛晖刘晓健
王奇峰,徐清华,陈金影,钱琛晖,刘晓健,杜 祥
1.复旦大学附属肿瘤医院病理科,复旦大学上海医学院肿瘤学系,上海 200032;
2.杭州可帮基因科技有限公司,浙江 杭州 311188;
3.复旦大学附属肿瘤医院化疗科,复旦大学上海医学院肿瘤学系,上海 200032
一种新型肿瘤组织起源分子标志物的建立与评价
王奇峰1,徐清华2,陈金影2,钱琛晖2,刘晓健3,杜 祥1
1.复旦大学附属肿瘤医院病理科,复旦大学上海医学院肿瘤学系,上海 200032;
2.杭州可帮基因科技有限公司,浙江 杭州 311188;
3.复旦大学附属肿瘤医院化疗科,复旦大学上海医学院肿瘤学系,上海 200032

杜 祥,复旦大学附属肿瘤医院主任医师、教授、博士生导师。现任中国抗癌协会肿瘤病理专业委员会候任主委、中国研究型医院学会病理学专业委员会主任委员、中国医师协会病理科医师分会副会长、中华医学会病理分会常委;中国医学装备协会病理装备技术专业委员会副主任委员、国家标准委全国生物样本技术委员会副主任委员、中国医药生物技术协会组织样本库分会副主任委员。从事肿瘤的病理诊断工作26年,围绕恶性肿瘤发生机制、分子分型及诊断预后标志物等方面开展研究。2007年至今在国内外医学专业期刊上以通信作者发表论文48篇;多次获得中国抗癌协会科技奖、教育部科技进步奖和上海市科技进步奖;承担国家自然科学基金、上海市基础研究重大项目和重点项目及其他各类科研项目10余项,目前作为课题第一负责人承担国家自然科学基金、上海市基础研究重点项目、卫生部临床重点专科项目等在研课题7项。
背景与目的:原发灶不明恶性肿瘤是一类转移性肿瘤的统称,在诊断时无法找到原发位点,约占所有恶性肿瘤的5%~10%。明确肿瘤的组织起源对于患者的诊断和治疗具有重要意义。方法:整合ArrayExpress和Gene Expression Omnibus数据库中肿瘤类型明确的样本数据,构建涵盖22种常见肿瘤类型、5 800例样本的基因表达谱数据库;通过支持向量机递归特征消除算法筛选组织特异性基因,建立肿瘤分类模型;采用实时定量聚合酶链反应(real-time quantitative polymerase chain reaction,RTQ-PCR)检测石蜡包埋肿瘤组织中基因的表达水平,并将基因分型结果与病理诊断结果进行比较。结果:基于肿瘤基因表达谱大数据,筛选出96个组织特异性基因,其中包含常见的肿瘤相关基因,如钙黏蛋白1(cadherin 1,CDH1)、激肽释放酶相关酶3(kallikreinrelated peptidase 3,KLK3)和表皮生长因子受体(epidermal growth factor receptor,EGFR)等。在206例石蜡包埋组织样本中,182例的基因分型结果与病理诊断结果一致,准确率达到88.4%(95%CI:83.2%~92.4%)。结论:96基因RTQ-PCR检测对22种常见肿瘤类型具有较好的分类性能,可作为临床和病理诊断的辅助工具。
原发灶不明恶性肿瘤;肿瘤组织起源;基因表达谱;实时定量聚合酶链反应;免疫组化
原发灶不明恶性肿瘤(cancer of unknown primary,CUP)是一类经病理学诊断确诊为转移性恶性,但是通过详细评估未能明确原发位点的异质性肿瘤[1]。据统计,CUP约占全部肿瘤病例的5%~10%[2],居常见恶性肿瘤的第8位[3],死亡率则高居第4位[4]。一项荟萃研究显示,原发灶不明恶性肿瘤患者接受化疗后中位生存时间为4.5个月,1年生存率为20%,5年生存率为4.7%[5],CUP患者的预后很大程度上取决于原发肿瘤的生物学特性,因此找出肿瘤的组织起源,采取有针对性的治疗,对于改善患者预后具有重要意义。
CUP的临床评估包括病史询问、体格检查、实验室检查、内镜及影像学检查等。PET/ CT是目前最有效的影像学识别CUP原发位点的工具,诊断率为24%~53%[6]。病理检查对寻找原发灶具有重要的价值。对于少数具有原发肿瘤典型结构的转移性肿瘤如肾透明细胞癌、甲状腺滤泡状腺癌等,病理医师通过形态学观察后容易判断出原发灶;对大多数不具备典型结构的转移性肿瘤,也可采用免疫组织化学标志物推测肿瘤细胞类型和组织来源。然而即使通过详尽的临床、影像和病理检查,仍有20%~50%的患者无法找出原发灶[7]。
近年来,随着生物技术的飞速发展,研究人员可同时检测肿瘤组织中成千上万个基因的表达水平,从中发现与肿瘤组织起源相关的基因及特定的表达模式。转移灶肿瘤的基因表达谱与转移部位组织的基因表达谱存在差异,而与其原发部位组织的基因表达谱更相似,提示肿瘤在其发生、发展和转移的过程中,始终保留其组织起源的基因表达特征。根据这一原理,Xu等[8]通过基因表达谱分析,构建了1项包含154个组织特异性基因的分子标志物,可用于判定22种常见肿瘤类型和组织起源。在此基础之上,本研究进一步将组织特异基因的数目由154个减少到96个,采用实时定量聚合酶链反应(real-time quantitative polymerase chain reaction,RTQ-PCR)检测96基因在4%甲醛溶液固定石蜡包埋(formalin-fixed paraffinembedded,FFPE)样本中的特异性表达,并根据RTQ-PCR技术特点对96基因分子标志物进行优化,以利于实现该成果向临床应用的转化。
1 资料和方法
1.1 肿瘤基因表达谱数据库
从欧洲生物信息学研究所的ArrayExpress数据库和美国生物技术信息中心的Gene ExpressionOmnibus数据库中选取肿瘤类型明确的样本,共计5 800例。对样本的临床信息进行标准化和规范化处理后构建包含22种肿瘤类型、涵盖95%以上实体肿瘤的肿瘤基因表达谱数据库。基因表达谱分析采用美国Affymetrix公司生产的、多种规格的人类全基因组芯片,包括GeneChip® Human Genome U133A、U133A 2.0和U133 plus 2.0芯片。首先,采用Single Channel Array Normalization算法对基因芯片的原始数据进行预处理。Single Channel Array Normalization算法能够实现对单个样本的数据结构解析,去除由引物、探针引起的背景噪声对生物学信号的干扰,因此相比于其他需多个样本数据同时解析的均一化算法,更加适用于个体化分子诊断的工作流程[9]。其后,采用BrainArray Resource提供的基因组信息注释不同型号芯片的探针,并将其统一映射到Entrez Gene ID[10]。
1.2 临床样本
标本源自复旦大学附属肿瘤医院和江苏省常州市第一人民医院在2012—2016年收治的患者。其中,男性100例(48.5%),女性106例(51.5%);年龄范围为6~82岁,平均年龄54岁。所有样本经病理诊断明确原发位点,分属22种常见肿瘤类型。其中,肾上腺肿瘤8例,脑肿瘤12例,乳腺癌9例,宫颈癌8例,结直肠癌10例,子宫内膜癌10例,胃及食管癌17例,生殖细胞肿瘤9例,头颈部肿瘤9例,肾癌8例,肝胆肿瘤8例,肺癌9例,淋巴瘤9例,黑素瘤9例,间皮瘤9例,神经内分泌肿瘤10例,卵巢癌9例,胰腺癌8例,前列腺癌8例,肉瘤11例,甲状腺癌8例,尿路上皮癌8例。
1.3 仪器与试剂
RecoverAll™ Total Nucleic Acid Isolation Kit for FFPE抽提试剂盒购自美国Ambion公司;High-Capacity cDNA Reverse Transcription Kit反转录试剂盒和Master Mix试剂购自美国Applied Biosystems公司。Taqman™ MGB探针和引物由美国Invitrogen公司设计合成。采用Applied Biosystems®7500荧光定量PCR系统进行检测。
1.4 实验方法
每例标本的组织蜡块连续切6张10 μm厚的切片。按RNA抽提试剂盒所述方法提取样本总RNA,收集总RNA提取液40 μL。用微量紫外可见光分光光度计测定浓度和纯度。将总RNA反转录成cDNA,反转录完成后,-20 ℃保存备用。
在已加入引物探针预混液4.0 μL的96孔板上对反转录后的cDNA进行扩增,PCR反应体系20.0 μL:Master Mix 10.0 μL,cDNA模板+H20 6.0 μL。将96孔板置于ABI7500荧光定量PCR仪。反应条件:95 ℃预变性10 min;95 ℃变性15 s,60 ℃退火延伸1 min,40个循环。信号采集设在延伸步骤。
1.5 数据分析
数据的读取、存储、分析和处理主要采用R统计语言和Bioconductor项目开发的程序包[11-13]。采用支持向量机递归特征消除算法(support vector machine-recursive feature elimination,SVM-RFE)进行特征选择和分类建模[14]。针对多类别分类问题,采用“一对一”的分析策略,即在每两类之间训练1个SVM分类器。因此针对22种肿瘤类型的分类问题,训练阶段共构造231个两类分类器,每个分类器是取任意2个类别的数据进行训练。对于第i类和第j类之间的训练,需要解决下面的两类分类问题:
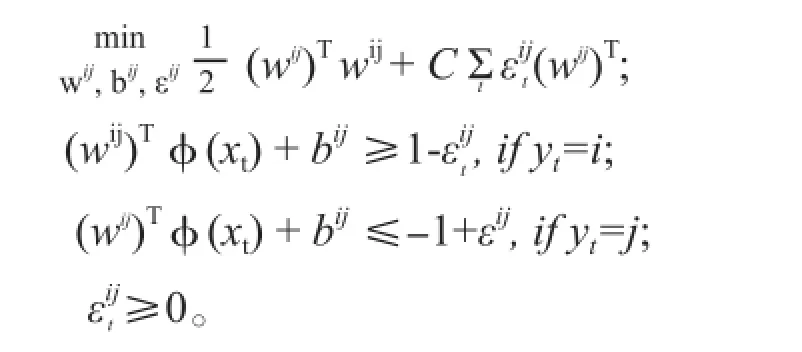
测试阶段确定样本属于哪一类,选择最常用的“最大投票法”,即每个两类分类器都对样本的类别进行判断,采用投票机制为其相应的类别投上一票,最后得票最多的类即是该未知样本的所属类。对待测样本x进行分类时,判断符号函数:
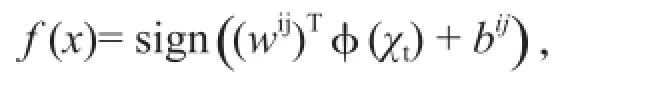
若x属于第i类,则第i类的票数加1,反之第j类加1。待测样本x属于最后票数最多的那一类。为更好地展示待测样本与所有22种肿瘤类型的接近程度,我们进一步采用sigmoid-fitting方法[15-16],计算待测样本属于各种肿瘤类型的概率,并将其命名为相似度分数。
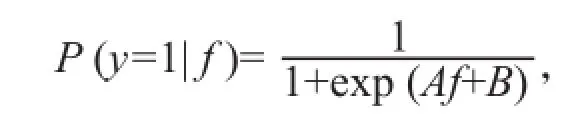
待测样本与每种肿瘤类型的相似度分数在0(低概率)到100(高概率)之间变化。22种肿瘤类型的相似度分数之和为100。根据相似度最高原则判定该样本最可能的肿瘤类型。96基因RTQPCR检测及数据分析过程见图1。
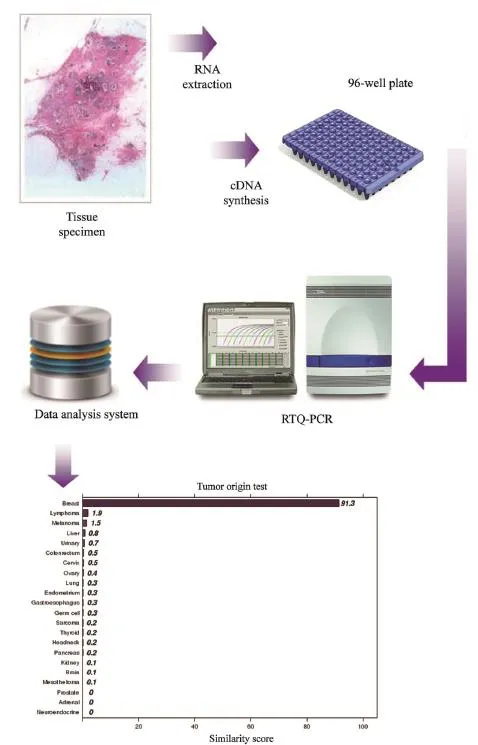
图 1 96基因RTQ-PCR检测及数据分析过程Fig. 1 96-gene RTQ-PCR assay testing workflow
1.6 分类模型性能评估
将9 6基因分型结果与病理诊断结果进行比对。以病理诊断为金标准,根据表1计算灵敏度、特异度、诊断符合率及9 5%C I[17]。

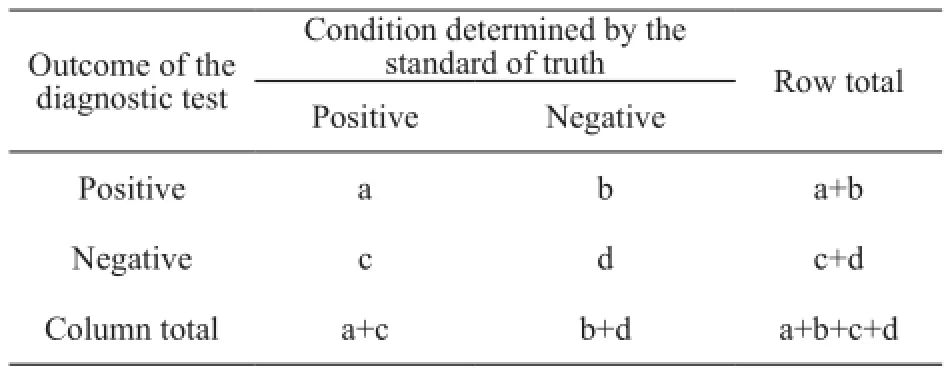
表 1 灵敏度、特异度和诊断符合率计算Tab. 1 Sensitivity, specificity and accuracy
2 结 果
2.1 肿瘤基因表达谱数据库的构建
构建肿瘤基因表达谱数据库时优先考虑3个方面特性:① 应涵盖尽可能多的肿瘤类型;② 对于具有明显异质性的肿瘤类型,应包含尽可能多的组织亚型;③ 纳入转移性肿瘤和分化差的样本,从而尽可能真实地评估分子标志物识别CUP样本组织起源的性能。基于上述3点,我们从ArrayExpress和Gene Expression Omnibus数据库中收集了囊括22种肿瘤类型、5 800例肿瘤样本的基因表达谱数据。22种肿瘤类型包括肾上腺肿瘤、脑肿瘤、乳腺癌、宫颈癌、结直肠癌、子宫内膜癌、胃及食管癌、生殖细胞肿瘤、头颈部肿瘤、肾癌、肝胆肿瘤、肺癌、淋巴瘤、黑素瘤、间皮瘤、神经内分泌肿瘤、卵巢癌、胰腺癌、前列腺癌、肉瘤、甲状腺癌和尿路上皮癌等。每种肿瘤的样本量从55例到542例不等(表2)。所有的5 800例样本数据都作为训练集,用于后续组织特异基因的筛选及分类模型的建立。
2.2 基因筛选与功能注释
以前期筛选的154个基因[8]为基础,我们进一步运用SVM-RFE算法,针对每一种肿瘤类型:① 评估每个基因对区分该肿瘤类型的贡献值;② 选取对该肿瘤类型贡献最大的13个基因作为特异表达基因;③ 对22种肿瘤类型重复上述步骤。除去22组基因之间存在的交叉重叠,最终筛选得到96个组织特异基因(表3)。值得注意的是,96个基因中包含一些文献中已报道的常见肿瘤相关基因,例如激肽释放酶相关酶3(kallikrein-related peptidase 3,KLK3)所编码的前列腺特异抗原,是诊断和监测前列腺癌最重要的肿瘤标志物;另外,表皮生长因子受体(epidermal growth factor receptor,EGFR)基因在脑肿瘤、结直肠癌、肺癌、食管癌、宫颈癌和肉瘤等多种肿瘤中特异表达[18-23];钙黏蛋白1(cadherin 1,CDH1)和血管内皮生长因子A(vascular endothelial growth factor A,VEGFA)则是结直肠癌、胃癌和肝癌重要的分子标志物[24-26]。采用GeneCodis Bioinformatics Tool对96个基因进行Kyoto Encyclopedia of Genes and Genomes(KEGG)通路富集分析,进而揭示特征基因所反映的生物学意义。特征基因在细胞因子-受体相互作用、蛋白质消化和吸收及PPAR信号通路等生物学通路中显著富集(表4)。2.3 96基因模型的性能验证
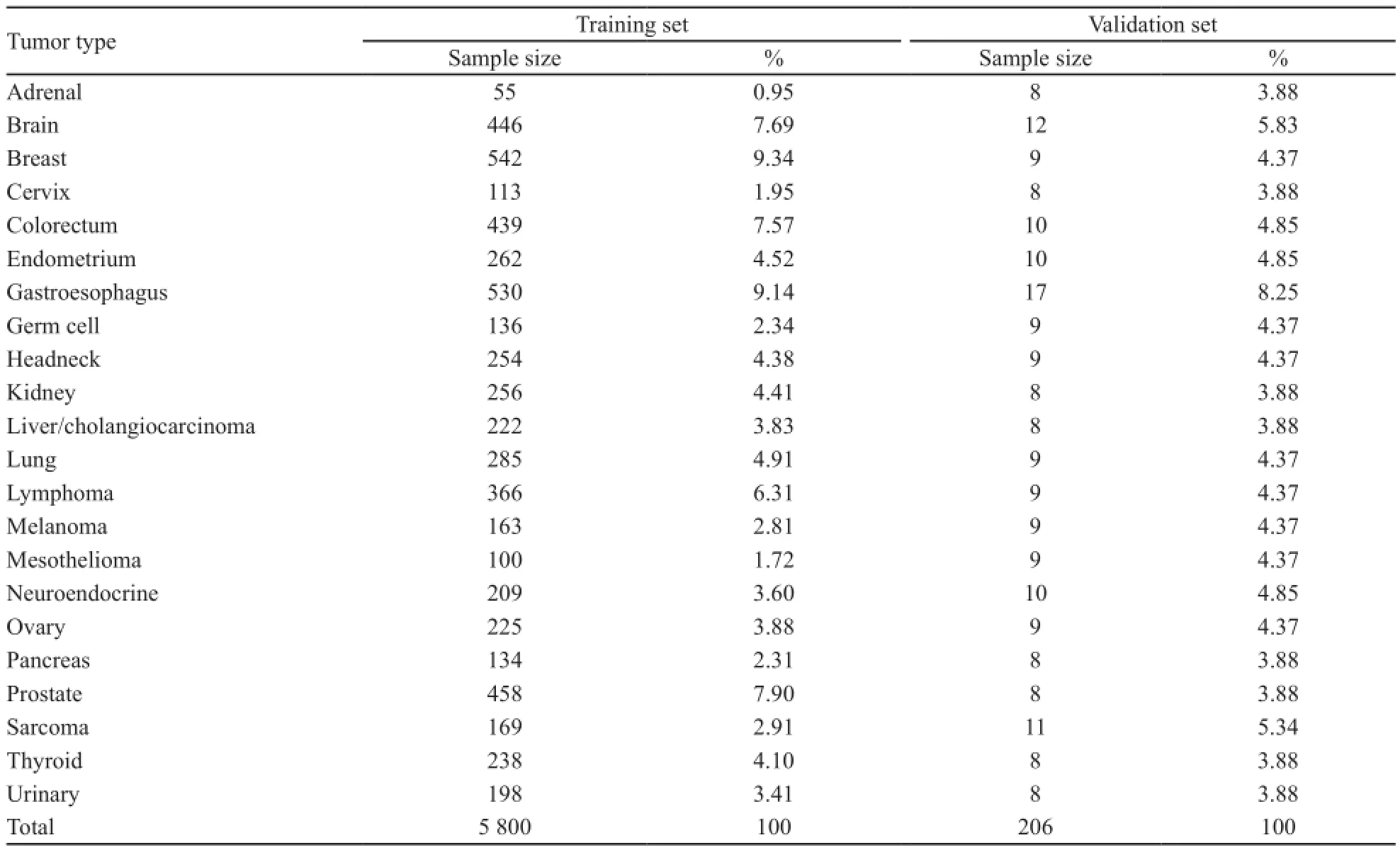
表 2 样本临床信息Tab. 2 Summary of sample information
表 3 96基因列表Tab. 3 List of selected 96 genes

续表3

续表3
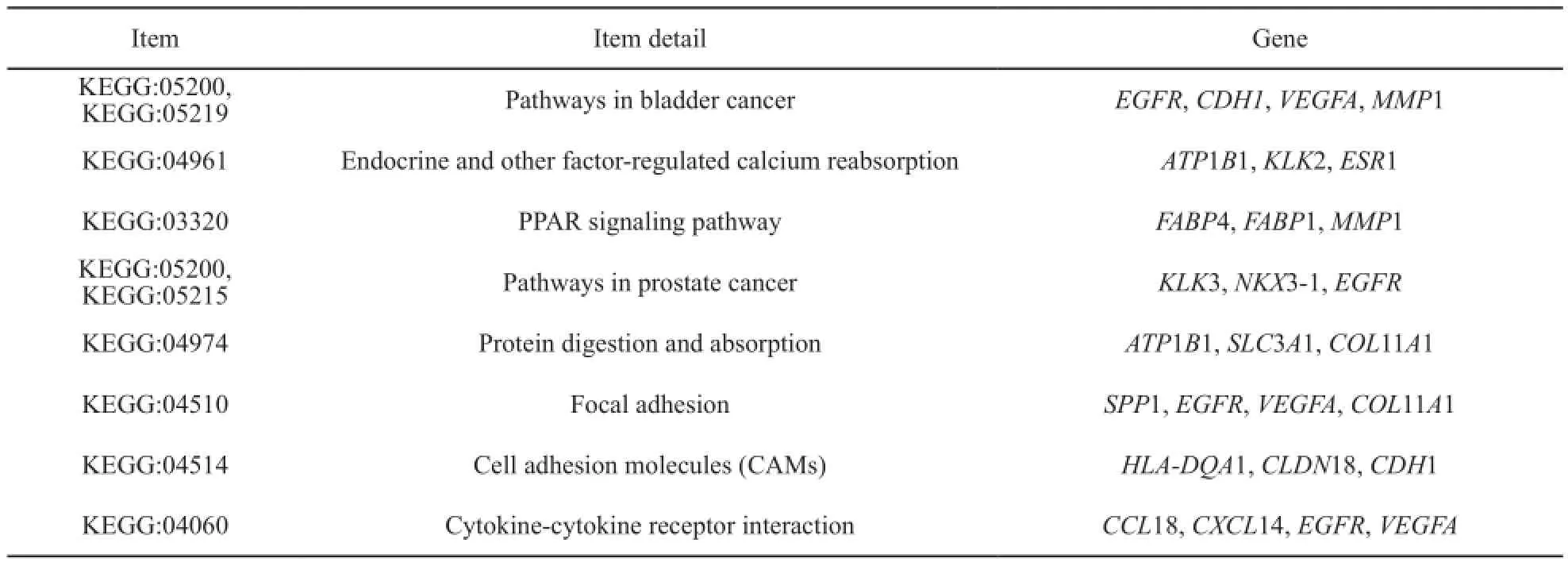
表 4 96基因KEGG通路富集分析Tab. 4 The top KEGG pathways enriched in the 96-gene list
首先,在训练集中评估96基因分类模型判别各类肿瘤的准确率。采用“留一法交叉验证法”,即在数据集中每次仅保留一例样本作为测试样本,其余样本均用作训练样本,重复该过程,直到所有的样本均被用作测试样本为止。96基因模型的整体准确率为95.3%(95%CI:94.4%~95.5%)。训练集的验证结果提示,96基因模型对于各类肿瘤具有较好的区分能力,但由于存在数据过度拟合的风险,因此,需要将96基因模型进一步应用于临床样本进行测试。
通过RTQ-PCR检测FFPE样本中96个肿瘤组织特异性基因的表达模式,并由SVM分类模型判定样本的肿瘤类型。在206例FFPE样本中,182例样本的基因检测分析结果与组织病理诊断结果一致,因此诊断符合率为88.4%(95%CI: 83.2%~92.4%)。96基因模型对于22种肿瘤的分析结果见表5,其中肾上腺肿瘤、脑肿瘤、乳腺癌、结直肠癌、肾癌、肝胆肿瘤、淋巴瘤、卵巢癌、前列腺癌和甲状腺癌的灵敏度达100%;头颈部肿瘤、间皮瘤、胰腺癌、神经内分泌肿瘤、尿路上皮癌、肾上腺肿瘤、肺癌、肾癌、淋巴瘤、恶性黑素瘤和甲状腺癌的特异度达100%。
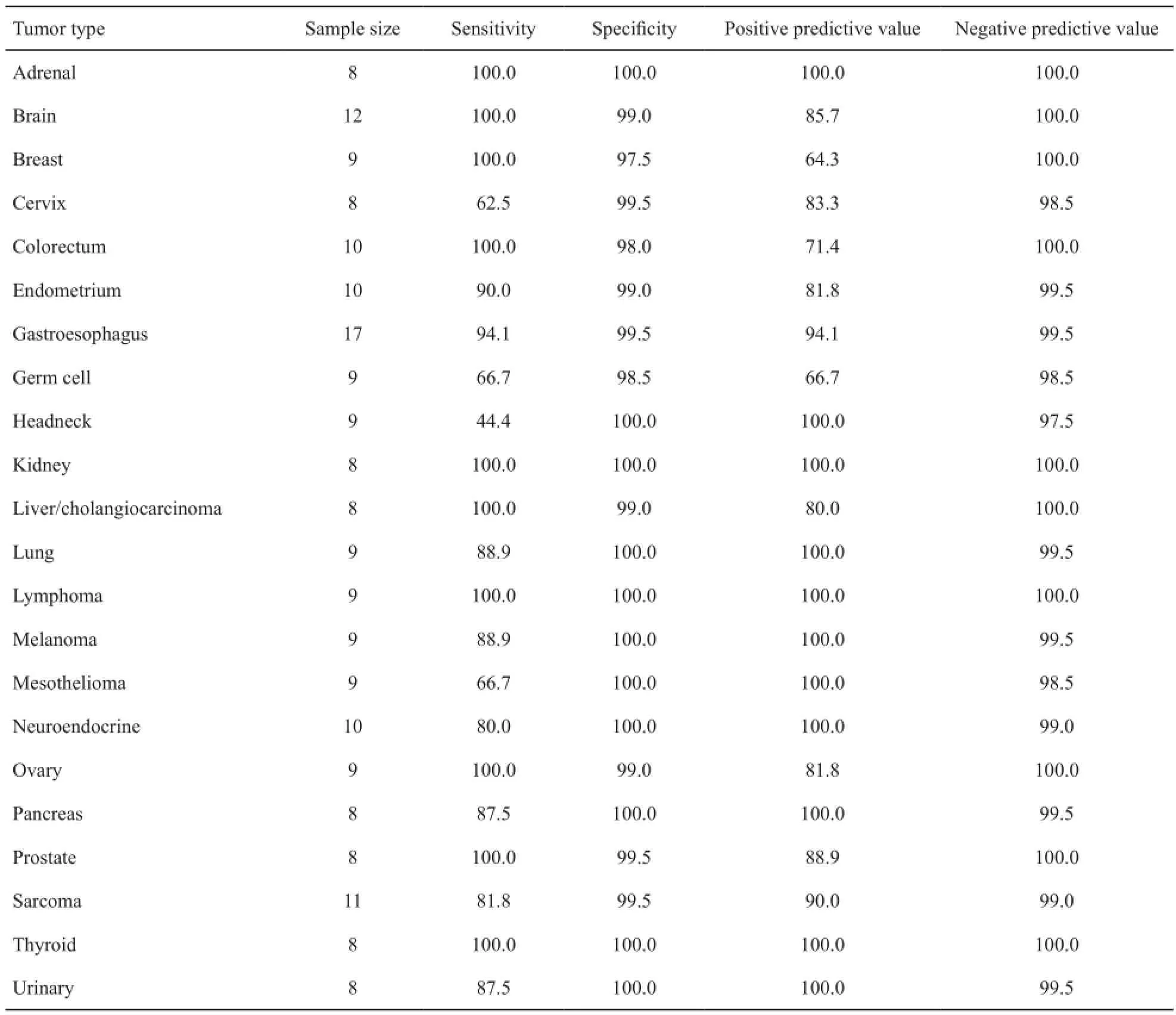
表 5 96基因标志物性能验证Tab. 5 Performance characteristics of the 96-gene expression signature(%)
根据Chen等[27]公布的2015年统计数据,在我国最常见十大肿瘤中,96基因模型的诊断符合率为93.3%(95%CI:85.4%~97.2%)。在男性最常见十大肿瘤中,准确率为95.5%(95%CI:88.2%~98.6%);在女性最常见十大肿瘤中,准确率为93.2%(95%CI: 85.2%~97.2%,表6、7、8)。
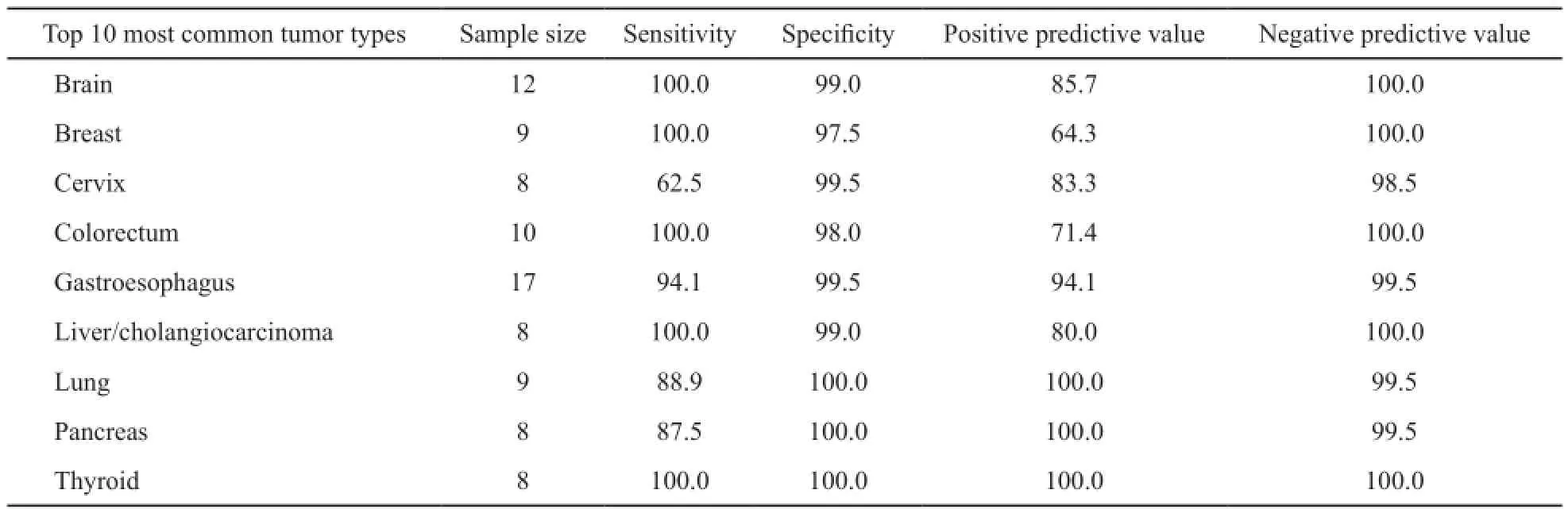
表 6 96基因标志物在最常见十大肿瘤中的性能Tab. 6 Performance characteristics of the 96-gene expression signature in 10 most frequent cancers(%)
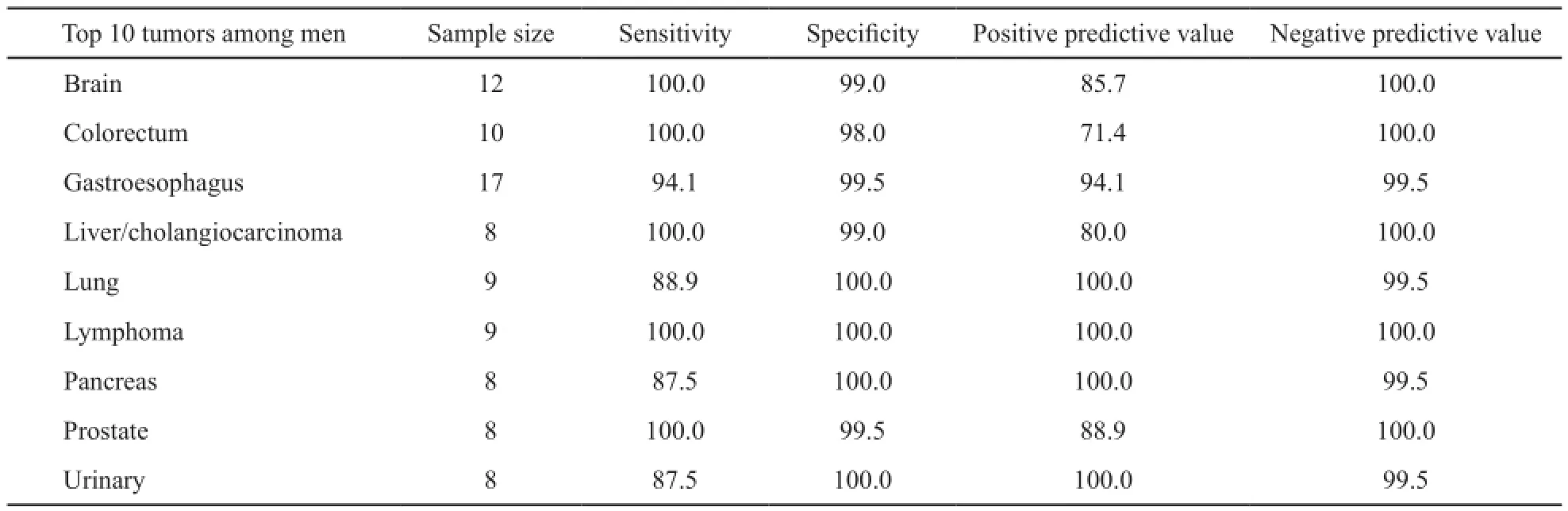
表 7 96基因标志物在男性最常见十大肿瘤中的性能Tab. 7 Performance characteristics of the 96-gene expression signature in 10 most common cancers in males(%)
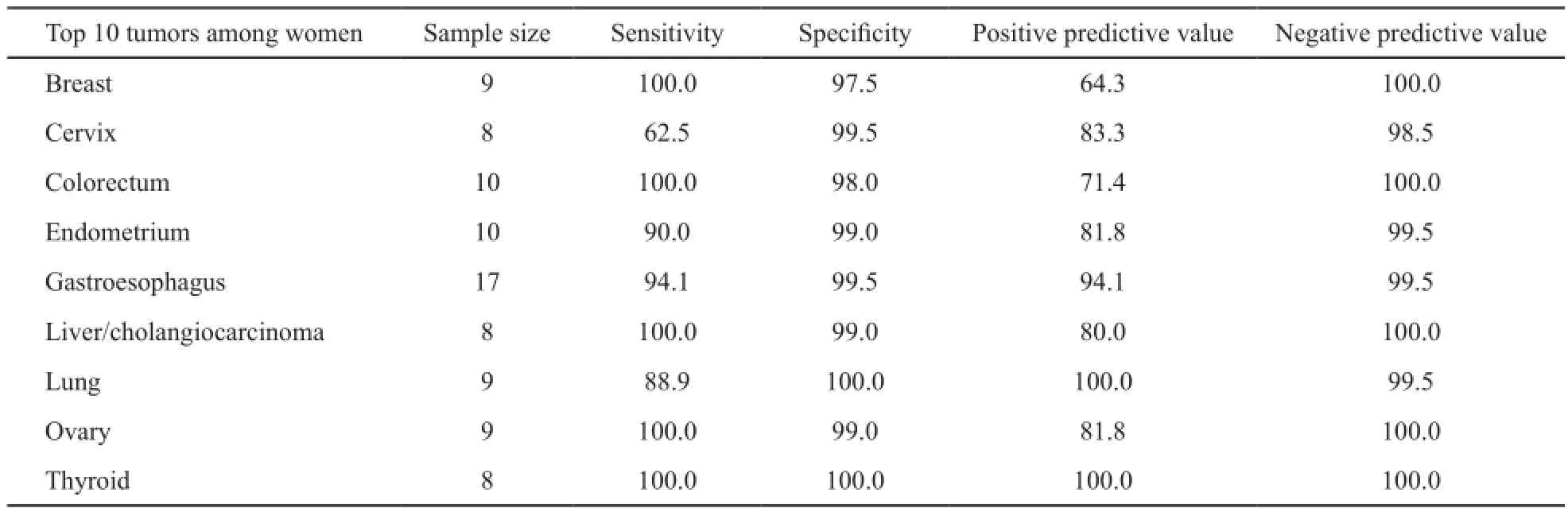
表 8 96基因标志物在女性最常见十大肿瘤中的性能Tab. 8 Performance characteristics of the 96-gene expression signature in 10 most common cancers in females(%)
3 讨 论
近年来,飞速发展的基因组、转录组、蛋白质组和代谢组等组学技术产生了海量的研究数据。生物医学大数据与生物信息学的有机结合对从分子水平阐明肿瘤本质及准确划分肿瘤类型起到重要的辅助作用。国内外多项研究证实,分子标志物可用于识别肿瘤组织起源。Talantov等[28]通过RTQ-PCR测定10个基因的表达水平,判别原发位点是否来源于肺、乳腺、结肠、卵巢、胰腺和前列腺。在260个已知原发位点的转移性肿瘤样本中准确率为78%。Ma等[29]采用RTQ-PCR检测92个基因的表达水平,可识别32种肿瘤的原发部位,该方法准确率为87%。Rosenfeld等[30]则通过检测48个microRNAs的表达水平,识别22种肿瘤的组织起源,准确率为89%。Park等[31]采用10个免疫组织化学标志物组合鉴别转移性肿瘤的原发位点,准确率为75%,提示分子标志物较免疫组织化学标志物具有更高的准确率。相比于影像学和组织病理诊断方法,分子标志物检测具有灵敏度和特异度高、结果判读客观等优势,在欧美一些发达国家已作为辅助手段应用于CUP原发位点的诊断[32-33]。
本研究整合ArrayExpress和Gene Expression Omnibus数据库中肿瘤类型明确的生物芯片数据,构建涵盖22大类肿瘤、5 800例样本的基因表达谱数据库;从中筛选出96个组织特异性基因,建立肿瘤分类模型;并通过优化RNA提取方法及PCR探针引物设计,实现在FFPE样本中定量检测上述基因的表达水平。在206例FFPE样本中,182例的基因分型结果与病理诊断结果一致,分类准确率达88.4%。这一分类性能与基于生物芯片研究的结果具有较好的一致性[8],因此验证了多基因分子标志物从生物芯片平台转化到RTQ-PCR平台的可行性。在我国男性和女性最常见十大肿瘤中,96基因模型的分类准确率分别达到95.5%和93.2%,提示该检测在国内具有较好的应用前景。
然而,本研究仍存在一定的局限性。96基因模型对于部分肿瘤类型的灵敏度较低,如头颈部肿瘤为44.4%、宫颈癌为62.5%。这可能是由于特定肿瘤类型自身存在较大的异质性,如头颈部肿瘤;也可能由于部分肿瘤如宫颈癌与起源于子宫内膜的癌具有相同的胚胎起源,因此呈现出相似的组织形态和基因表型。上述肿瘤类型较低的准确率在其他研究中也有报道[34]。尽管在本研究中包含了相当比例的分化不良肿瘤样本(77.5%),但其主要来源于肿瘤原发灶,后续研究应纳入更多转移性肿瘤样本进行验证。
综上所述,本研究在前期工作的基础上,实现了肿瘤组织起源分子标志物从生物芯片到RTQ-PCR技术平台的转化,因此更有利于该项成果的临床应用。本研究显示,96基因RTQPCR检测对于不同肿瘤的FFPE样本具有较好的判别能力,展现出其在CUP患者临床诊断中的潜在价值。后续工作需进一步结合临床实践,通过前瞻性研究设计,比较基因检测指导下的治疗与经验性治疗的疗效,进而评估基因检测对于CUP患者治疗和预后的意义。
[1] STELLA G M, SENETTA R, CASSENTI A, et al. Cancers of unknown primary origin: current perspectives and future therapeutic strategies[J]. J Transl Med, 2012, 10: 12.
[2] 张延龄. 原发灶不明的肿瘤患者的处理(文献综述)[J].国外医学外科学分册, 2002, 29(5): 282-285.
[3] PAVLIDIS N, FIZAZI K. Cancer of unknown primary (CUP)[J]. Crit Rev Oncol Hematol, 2005, 54(3): 243-250.
[4] KAMPOSIORAS K, PENTHEROUDAKIS G, PAVLIDIS N. Exploring the biology of cancer of unknown primary: breakthroughs and drawbacks[J]. Eur J Clin Invest, 2013,43(5): 491-500.
[5] RICHARDSON A, WAGLAND R, FOSTER R, et al. Uncertainty and anxiety in the cancer of unknown primary patient journey: a multiperspective qualitative study[J]. BMJ Support Palliat Care, 2015, 5(4): 366-372.
[6] RESKE S N, KOTZERKE J. FDG-PET for clinical use. Results of the 3rd German Interdisciplinary Consensus Conference, “Onko-PET Ⅲ”, 21 July and 19 September 2000[J]. Eur J Nucl Med, 2001, 28(11): 1707-1723.
[7] 潘宏铭, 郑 宇. 原发灶不明转移癌的诊断与治疗进展[C]/中国肿瘤内科进展 中国肿瘤医师教育. 2014.
[8] XU Q, CHEN J, NI S, et al. Pan-cancer transcriptome analysis reveals a gene expression signature for the identification of tumor tissue origin[J]. Mod Pathol, 2016, 29(6): 546-556.
[9] PICCOLO S R, SUN Y, CAMPBELL J D, et al. A singlesample microarray normalization method to facilitate personalized-medicine workflows[J]. Genomics, 2012,100(6): 337-344.
[10] DAI M, WANG P, BOYD A D, et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data[J]. Nucleic Acids Res, 2005, 33(20): e175.
[11] IHAKA R, GENTLEMAN R. R: a language for data analysis and graphics[J]. J Comput Graph Stat, 1996, 5(3): 299-314.
[12] REIMERS M, CAREY V J. Bioconductor: an open source framework for bioinformatics and computational biology[J]. Methods Enzymol, 2006, 411(411): 119-134.
[13] CHANG C, LIN C. LIBSVM: A library for support vector machines[J]. ACM Trans Intell Syst Technol, 2011, 2(3): 389-396.
[14] GUYON I, WESTON J, BARNHILL S, et al. Gene selection for cancer classification using support vector machines[J]. Mach Learn, 2001, 46(1-3): 389-422.
[15] WU T, LIN C, WENG R C. Probability estimates for multiclass classification by pairwise coupling[J]. J Mach Learn Res, 2004, 5(4): 975-1005.
[16] PLATT J C. Probabilistic outputs for support vector machines and comparisons to regularized likelihood methods[J]. Advances in Large Margin Classifiers, 2000, 10: 61-74.
[17] 李 康. 《医学统计学》[M]. 北京: 人民卫生出版社,2013: 216-218.
[18] DEVARAKONDA S, MORGENSZTERN D, GOVINDAN R. Genomic alterations in lung adenocarcinoma[J]. Lancet Oncol, 2015, 16(7): e342-e351.
[19] FURNARI F B, CLOUGHESY T F, CAVENEE W K, et al. Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma[J]. Nat Rev Cancer, 2015, 15(5): 302-310.
[20] GIAMPIERI R, APRILE G, DEL P M, et al. Beyond RAS: the role of epidermal growth factor receptor (EGFR) and its network in the prediction of clinical outcome during anti-EGFR treatment in colorectal cancer patients[J]. Curr Drug Targets, 2014, 15(13): 1225-1230.
[21] LI J C, ZHAO Y H, WANG X Y, et al. Clinical significance of the expression of EGFR signaling pathway-related proteins in esophageal squamous cell carcinoma[J]. Tumour Biol,2014, 35(1): 651-657.
[22] LI Q, TANG Y, CHENG X, et al. EGFR protein expression and gene amplification in squamous intraepithelial lesions and squamous cell carcinomas of the cervix[J]. Int J Clin Exp Pathol, 2014, 7(2): 733-741.
[23] TENG H W, WANG H W, CHEN W M, et al. Prevalence and prognostic influence of genomic changes of EGFR pathway markers in synovial sarcoma[J]. J Surg Oncol, 2011,103(8): 773-781.
[24] JING H, DAI F, ZHAO C, et al. Association of genetic variants in and promoter hypermethylation of CDH1 with gastric cancer: a meta-analysis[J]. Medicine (Baltimore), 2014,93(19): e107.
[25] LI Y X, LU Y, LI C Y, et al. Role of CDH1 promoter methylation in colorectal carcinogenesis: a meta-analysis[J]. DNA Cell Biol, 2014, 33(7): 455-462.
[26] LIU F, LI H, CHANG H, et al. Identification of hepatocellular carcinoma-associated hub genes and pathways by integrated microarray analysis[J]. Tumori, 2015, 101(2): 206-214.
[27] CHEN W, ZHENG R, BAADE P D, et al. Cancer statistics in China, 2015[J]. CA Cancer J Clin, 2016, 66(2): 115-132.
[28] TALANTOV D, BADEN J, JATKOE T, et al. A quantitative reverse transcriptase-polymerase chain reaction assay to identify metastatic carcinoma tissue of origin[J]. J Mol Diagn, 2006, 8(3): 320-329.
[29] MA X J, PATEL R, WANG X, et al. Molecular classification of human cancers using a 92-gene real-time quantitative polymerase chain reaction assay[J]. Arch Pathol Lab Med,2006, 130(4): 465-473.
[30] ROSENFELD N, AHARONOV R, MEIRI E, et al. MicroRNAs accurately identify cancer tissue origin[J]. Nat Biotechnol,2008, 26(4): 462-469.
[31] PARK S Y, KIM B H, KIM J H, et al. Panels of immunohistochemical markers help determine primary sites of metastatic adenocarcinoma[J]. Arch Pathol Lab Med, 2007,131(10): 1561-1567.
[32] WEISS L M, CHU P, SCHROEDER B E, et al. Blinded comparator study of immunohistochemical analysis versus a 92-gene cancer classifier in the diagnosis of the primary site in metastatic tumors[J]. J Mol Diagn, 2013, 15(2): 263-269.
[33] PILLAI R, DEETER R, RIGL C T, et al. Validation and reproducibility of a microarray-based gene expression test for tumor identification in formalin-fixed, paraffin-embedded specimens[J]. J Mol Diagn, 2011, 13(1): 48-56.
[34] ERLANDER M G, MA X J, KESTY N C, et al. Performance and clinical evaluation of the 92-gene real-time PCR assay for tumor classification[J]. J Mol Diagn, 2011, 13(5): 493-503.
Identification and validation of a novel gene expression signature for diagnosing tumor tissue origin
WANG Qifeng1, XU Qinghua2, CHEN Jinying2, QIAN Chenhui2, LIU Xiaojian3, DU Xiang1
(1.Department of Pathology, Fudan University Shanghai Cancer Center, Department of Oncology, Shanghai Medical College, Fudan University, Shanghai 200032, China; 2.Canhelp Genomics Co., Ltd, Hangzhou 311188,Zhejiang Province, China; 3.Department of Chemotherapy, Fudan University Shanghai Cancer Center,Department of Oncology, Shanghai Medical College, Fudan University, Shanghai 200032, China)
Correspondence to: DU Xiang E-mail: dx2008cn@163.com
Background and purpose: Cancer of unknown primary (CUP) represents approximately 5%~10% of malignant neoplasms. For CUP patients, identification of tumor origin allows for more specific therapeutic regimens and improves outcomes. Methods: By retrieving the gene expression data from ArrayExpress and Gene Expression Omnibus data repositories, we established a comprehensive gene expression database of 5 800 tumor samples encompassing 22 main tumor types. The support vector machine-recursive feature elimination algorithm was used for feature selection and classification modelling. We further optimized the RNA isolation and real-time quantitative polymerase chain reaction (RTQ-PCR) methods for candidate gene expression profiling and applied the RTQ-PCR assays to a set of formalin-fixed, paraffin-embedded tumor samples. Results: Based on the pan-cancer transcriptome database, we identified a list of 96-tumor specific genes, including common tumor markers, such as cadherin 1 (CDH1), kallikrein-related peptidase 3 (KLK3), and epidermal growth factor receptor (EGFR). Furthermore, we successfully translated the microarray-based gene expression signature to the RTQ-PCR assays, which allowed an overall success rate of 88.4%(95%CI: 83.2%-92.4%) in classifying 22 different tumor types of 206 formalin-fixed, paraffin-embedded samples. Conclusion: The 96-gene RTQ-PCR assay represents a useful tool for accurately identifying tumor origins. The assay uses RTQ-PCR and routine formalin-fixed, paraffin-embedded samples, making it suitable for rapid clinical adoption.
Cancer of unknown primary; Tumor tissue origin; Gene expression profiling; Real-time quantitative polymerase chain reaction; Immunohistochemistry
10.19401/j.cnki.1007-3639.2016.10.001
R730.21
A
1007-3639(2016)10-0801-12
杜 祥 E-mail: dx2008cn@163.com
(2016-07-29
2016-09-08)
