一种来源于Burkholderia phytofirmans PsJN的ω-转氨酶的表达纯化及性质分析
2016-09-19杜允成董文玥姜进举陈启佳冯进辉吴洽庆朱敦明
杜允成,董文玥,姜进举,陈启佳,冯进辉,吴洽庆,朱敦明
一种来源于PsJN的ω-转氨酶的表达纯化及性质分析
杜允成1,2,董文玥1,姜进举1,2,陈启佳1,2,冯进辉1,吴洽庆1,朱敦明1
1 中国科学院天津工业生物技术研究所 工业酶国家工程实验室 天津 300308 2 中国科学院大学 北京 100049
ω-转氨酶 (ω-transaminase) 可以通过手性拆分和不对称合成的催化反应来获得光学纯的手性胺类化合物和非天然氨基酸,在医药中间体合成中是一种重要的生物催化剂。采用基因挖掘技术,在基因组数据库中获得一个来自伯克氏菌PsJN的ω-转氨酶基因 (),将该基因在大肠杆菌BL21 (DE3) 中克隆、表达,利用镍柱亲和层析将该酶 (HBP) 进行纯化并研究了其酶学性质和底物谱。结果表明,以β-苯丙氨酸 (β-Phe) 为氨基供体、丙酮酸为氨基受体,HBP具有较高的活力 (33.80 U/mg) 和立体选择性;其最适温度为40 ℃左右,最适pH在8.0–8.5之间。研究过程中,建立了一种简便快捷的紫外吸收法来检测β-Phe的脱氨反应,证明了该反应的热力学平衡性质。底物谱研究表明HBP可以以β-Phe及其衍生物为氨基供体。结果表明HBP能够有效地手性拆分-β-Phe及其衍生物,转化率在50%左右,ee>99%。
ω-转氨酶,伯克氏菌,芳香族β-氨基酸,手性拆分
转氨酶 (EC 2.6.1.X) 又叫氨基转移酶,是一种以磷酸吡哆醛 (PLP) 为辅酶,能够将氨基供体的氨基基团转移至氨基受体羰基位置的生物催化剂[1]。根据转氨酶作用的底物,转氨酶可以分为α-转氨酶和ω-转氨酶两大类。与α-转氨酶不同,ω-转氨酶催化的两个底物,至少有一个为非α-酮酸或者α-氨基酸 (图1)[2]。ω-转氨酶能够通过手性拆分或者不对称合成的方法催化合成光学纯的手性胺类化合物和非天然氨基酸。由于医药和农药工业中,大约有40%的生物活性物质都含有手性胺结构[1],因此,ω-转氨酶在有机合成中是一种非常重要的生物催化剂。

图1 ω-转氨酶催化的反应
上世纪50年代,实验证实了在哺乳动物中存在ω-转氨酶催化的化学反应[3]。1990年,Celgene公司在ω-转氨酶研究方面取得了重大突破,实现了ω-转氨酶催化手性胺类化合物的拆分[4],这项研究极大地促进了ω-转氨酶的研究。在过去的20多年中,ω-转氨酶催化合成手性胺类化合物及非天然氨基酸的研究广受关注并取得了巨大的进展[5-7]。1999年,Celgro公司以异丙胺和甲氧基异丙酮为底物,建立了ω-转氨酶催化()-甲氧基丙胺的生产路线[8];这是ω-转氨酶工业生产农用手性胺类药物中间体的代表。2010年,美国Codexis和Merck公司,对ω-转氨酶进行了11轮的突变,成功实现了酶法催化西他列汀 (治疗糖尿病药物) 的合成工艺,彻底取代了重金属铑催化的化学合成路线[9];该成果获得了2010年美国总统绿色化学奖,是生物催化工业应用的一个里程碑。
根据ω-转氨酶作用底物的立体构型,可将ω-转氨酶分为 ()-ω-转氨酶和 ()-ω-转氨酶。()-ω-转氨酶作用于 ()-型底物,()-ω-转氨酶作用于 ()-型底物。文献报道较多的是 ()-ω-转氨酶[5,10],其中研究最广泛、最透彻的是来自河流弧菌JS17的ω-转氨酶,该酶有两个底物结合口袋,大口袋能够接受底物的苯环或者羧基基团,小口袋对大于乙基的基团具有严格的空间位阻效应[11];由于小口袋限制,大大缩小了其应用范围,Nobili等通过分子改造扩大了这个酶的小口袋[12]。与 ()-ω-转氨酶相比,()-ω-转氨酶的数量屈指可数;直到2010年,Höhne等通过生物信息学分析,根据两个Motif对6 000多条标注为“L-分支链氨基酸转移酶”氨基酸序列进行逐条分析,最终获得了21条可能的 ()-ω-转氨酶序列,实验确定其中17条为 ()-ω-转氨酶[13];随后,我们实验室将两个Motif合并,又成功挖掘出5个 ()-ω-转氨酶[14]。()-ω-转氨酶逐步丰富起来。
ω-转氨酶不仅可以催化手性胺类化合物,还可以实现β-氨基酸的手性合成。虽然已有多种酶法合成β-氨基酸[15-18],但由于ω-转氨酶拥有反应快速、底物谱广泛以及不需要辅因子再生等优势,利用ω-转氨酶合成β-氨基酸是一种更加理想的选择。目前,能够作用于芳香族β-氨基酸的ω-转氨酶相对较少[19-23],寻找新的ω-转氨酶来催化获得光学纯的芳香族β-氨基酸具有重要意义。本文通过基因挖掘技术,获得了一种来自PsJN的ω-转氨酶,实现了芳香族β-氨基酸的手性拆分。
1 材料与方法
1.1 材料
1.1.1 质粒、菌株及主要试剂
pET-32a(+) 质粒购自Novagen公司;蛋白胨和酵母提取物均来自BD公司;Bradford BCA蛋白浓度试剂盒购自康为世纪公司;考马斯亮蓝R250购自Solarbio公司。氨基供体底物,-β-Phe (Alfa Aeser)、()-β-Phe (百灵威科技有限公司),-3-氨基-3-(4-氟苯基) 丙酸 (国药集团化学试剂有限公司)、-3-氨基- 3-(4-甲氧基苯基) 丙酸 (上海韶远化学科技有限公司)、-3-氨基-3-(4-甲基苯基) 丙酸 (国药集团化学试剂有限公司)、-3-氨基-3-(4-硝基苯基) 丙酸 (sigma公司)、-3-氨基-3-(4-羟基苯基) 丙酸 (上海韶远化学科技有限公司)、-3-氨基-4-甲基戊酸 (阿拉丁)、()-苯乙胺 (Alfa Aesar)、-2-戊胺 (TCI);氨基受体底物丙酮酸 (Alfa Aesar)、α-酮戊二酸 (国药集团化学试剂有限公司)、丙酮酸乙酯 (Alfa Aesar)、苯甲醛 (Alfa Aesar)、乙醛酸 (百灵威科技有限公司)、苯乙酮 (Alfa Aesar)、其他氨基受体 (sigma公司)。
1.1.2 主要仪器
高压匀浆破碎仪购自德国APV公司;RC6+高速冷冻离心机购自Thermo公司;凝胶成像系统购自BIO-RAD公司;ÄKTA purifier 10,所用层析柱均为GE公司;高效液相色谱仪 (HPLC 1260),购自Agilent公司;酶标仪 (SpectraMax M2e)。
1.2 基因挖掘
通过文献调研,发现来自根瘤菌属sp. LUK的 ()-立体选择性ω-转氨酶能够以β-Phe为氨基供体,以丙酮酸、草酰乙酸和α-酮戊二酸为氨基受体进行反应[20],表明这个酶能够催化芳香族β-氨基酸,并且有比较宽的氨基受体底物谱。因此,以来自sp. LUK的ω-转氨酶蛋白序列(EF127643.1) 为模板,在NCBI(http://blast. ncbi.nlm.nih.gov/Blast.cgi) 中进行Blastp (默认设置) 搜索,来挖掘能够作用于芳香族β-氨基酸的ω-转氨酶。从Blast结果列表中选择了一条来自PsJN,标注为“class III aminotransferase”的候选蛋白序列 (YP_001890102.1,将该蛋白序列在N端加上His6后命名为HBP),与已经报道的能够作用于芳香族β-氨基酸的ω-转氨酶(分别来自sp. LΜK[20]胞菌属sp. JS666[21]和争论贪噬菌属[22]、伯克氏菌属[23]) 进行Pr-Blast,HBP与之同源性都较高,分别为51%、55%、59%、83%。通过在线的jcat密码子优化程序 (http://www.jcat.de),对HBP的原始基因序列进行了针对(K12) 表达的密码子优化,在N端加上His6标签,目的基因直接合成至pET-32a原核表达载体的Ⅰ (691 bp处) 与HⅠ酶切位点之间。
1.3 目的蛋白的表达纯化
工程菌构建:将重组载体pET-32a-转化至BL21 (DE3) 感受态细胞中。
诱导表达:将工程菌接种到装有LB液体培养基 (Amp含量100 µg/mL) 的三角瓶 (800 mL/瓶) 里,置于37 ℃的恒温培养摇床 (200 r/min) 中培养至对数期 (600为0.6–1.0),加入终浓度为0.1 mmol/L的IPTG,置于25 ℃、200 r/min的恒温培养摇床中诱导表达12 h。
收菌破胞:将诱导表达的菌液离心并收集菌体,用冰浴过的磷酸钠缓冲液 (50 mmol/L,pH 7.0) 清洗菌体两遍;之后将菌体用100 mL冰浴过的破胞缓冲液重悬,破胞缓冲液为磷酸钠缓冲液 (50 mmol/L,pH 7.0),其中添加有20 µmol/L磷酸吡哆醛 (PLP)、1 mmol/L的PMSF、0.01% (/) 的β-巯基乙醇、30 mmol/L咪唑和500 mmol/L氯化钠;重悬好的菌液用高压匀浆机 (提前预冷至4 ℃) 进行细胞破碎。
纯化透析:将细胞破碎液离心收集破菌上清液 (呈淡黄色),用0.45 μm的滤器过滤。纯化过程主要在ÄKTA蛋白质纯化仪上进行,所用层析柱为Ni-NTA亲和层析柱 (实验室自装10 mL手工柱),纯化用到的缓冲液主要为上样溶液A (pH 7.0的50 mmol/L的磷酸钠缓冲液,含有30 mmol/L咪唑和500 mmol/L的氯化钠) 和洗脱溶液B (pH 7.0的50 mmol/L磷酸钠缓冲液,含有500 mmol/L咪唑和500 mmol/L氯化钠),采用咪唑线性浓度对目的酶蛋白进行洗脱;得到纯酶后对其进行透析,透析缓冲液为含有20 µmol/L的PLP和0.01% (/) 的β-巯基乙醇的磷酸钠缓冲液 (50 mmol/L,pH 7.0),透析过程在4 ℃环境进行;透析完成之后,所得纯酶溶液加入10%的甘油后分装密封后保存在–80 ℃冰箱中冷藏备用。
1.4 酶学性质测定
1.4.1 反应体系
典型的反应体系:体积为1 mL的含有PLP (20 μmol/L)、氨基供体-β-Phe、氨基受体丙酮酸的缓冲液 (100 mmol/L),在水浴中孵育至与设定温度相同后,加入6.6 μg的HBP纯酶启动反应,反应5 min后,取100 μL的反应液加入200 μL的丙酮终止反应。丙酮中含有30 mmol/L的1-氟-2,4-二硝基苯基-5-L-丙氨酰胺 (氨基酸衍生剂,参见FDAA,Marfey’s Reagent说明书),然后加入40 μL的1 mol/L碳酸氢钠溶液,混匀,在40 ℃加热1 h,冷却至室温后,加入20 μL的2 mol/L盐酸溶液。衍生完成后,底物-β-Phe的减少和产物丙氨酸的生成通过HPLC进行测定。HPLC检测条件为:色谱柱为Eclipse XDB-C18 column (4.6 mm×150 mm,Agilent),流动相为三乙胺溶液 (50 mmol/L三乙胺,磷酸调节pH至3.0)∶乙腈=62.5∶37.5,等度洗脱,流速为0.8 mL/min,检测波长为 340 nm。
1.4.2 最适反应pH及其最适反应温度
最适反应pH的测定:采用典型的反应体系,氨基供体-β-Phe和氨基受体丙酮酸的浓度分别为20 mmol/L和10 mmol/L,反应温度为37 ℃,其余保持不变。以pH为变量,测定在一系列pH (6.0、7.0、7.5、8.0、8.5、9.0、10.0) 的条件下底物 (-β-Phe) 的减少量,通过比较不同pH条件下氨基底物的减少,来表征pH对该酶活力的影响。用到的 pH缓冲液 (浓度皆为100 mmol/L) 为醋酸钠缓冲液(pH 3.8–5.6)、磷酸钠缓冲液 (pH 5.8–7.6)、硼酸硼砂缓冲液 (pH 7.8–9.2) 和硼砂氢氧化钠缓冲液 (pH 9.3–10.1)。
最适反应温度的测定:采用典型的反应体系,反应pH为上述测定的最适反应pH (8.2),氨基供体-β-Phe和氨基受体丙酮酸的浓度分别为20 mmol/L和10 mmol/L,其余保持不变。以反应温度为变量,测定在一系列温度 (25 ℃、30 ℃、35 ℃、40 ℃、45 ℃、50 ℃、55 ℃、60 ℃) 条件下底物的减少量,通过比较不同反应温度条件下底物-β-Phe的减少,来表征反应温度对该酶活力的影响。
1.4.3 pH稳定性和温度稳定性
pH稳定性的测定:采用典型反应体系,氨基供体-β-Phe和氨基受体丙酮酸的浓度分别为20 mmol/L和10 mmol/L,反应温度为1.4.2测定的最适反应温度 (40 ℃),但是反应所用的酶分别在不同pH的缓冲液中进行酸碱处理。具体为:将多份酶分别置于pH为5.0、6.0、7.0、8.0、9.0和10.0的缓冲液中处理24 h (室温25 ℃),之后测定不同pH处理24 h之后的残留酶活力。通过比较各反应氨基供体底物-β-Phe的减少,便可以计算出经过不同pH处理之后的酶活损失情况。使用的pH缓冲液 (浓度皆为100 mmol/L) 为醋酸钠缓冲液 (pH 3.8–5.6)、磷酸钠缓冲液 (pH 5.8–7.6)、硼酸硼砂缓冲液 (pH 7.8–9.2) 和硼砂氢氧化钠缓冲液 (pH 9.3–10.1)。
温度稳定性的测定:采用典型反应体系 (氨基供体-β-Phe和氨基受体丙酮酸的浓度分别为20 mmol/L和10 mmol/L),反应温度和反应pH均为最适反应条件 (40 ℃、pH 8.2),但是反应所用的酶经过不同温度的热处理。具体为:将多份酶液 (50 mmol/L磷酸钠缓冲体系,pH 7.0) 分别置于30 ℃、 35 ℃、40 ℃、45 ℃、50 ℃、55℃和60 ℃温度下热处理20 min,测定不同温度下热处理之后的残留酶活力。通过比较各反应底物减少,来表示不同温度热处理之后的酶活损失情况。
1.4.4 动力学参数和底物抑制测定
HPLC检测该反应 (图2) 拥有精确度高的优点,已有报道中检测-β-Phe脱氨反应 (图2) 也都采用HPLC方法,但这种方法十分费时耗力。因此,本研究建立了一种紫外吸收法来测定该酶的酶活,并成功应用于动力学参数的测定中。

图2 HBP手性拆分rac-β-Phe
首先,由于产物C (3-羰基-3-苯基丙酸) 能够自发脱羧,是一种不稳定的β-酮酸类化合物[24],因此研究了温度对该化合物C稳定性的影响以及产物C的自发脱羧速率。温度对化合物C的影响:脂肪酶Nov435水解其酯而制备产物C (图3),水解完全后,将其置于不同的温度下处理2 h,此段时间内,产物C进行脱羧反应;2 h后,通过得到苯乙酮的量来计算其分解产率,分解产物苯乙酮是通过HPLC进行检测的。分解速率的确定:将产物C置于HPLC的样品盘中,每隔0.5 h检测一次样品,来测定此时苯乙酮的量;12 h后,100 ℃加热0.5 h,使产物C全部脱羧分解为苯乙酮,来确定苯乙酮的总量。HPLC检测条件为:色谱柱为Eclipse XDB-C18 column (4.6 mm×150 mm,Agilent),流动相为水∶乙腈=1∶1,等度洗脱,流速为1.0 ml/min,检测波长为205 nm。
其次,产物C有一种特殊的超级共轭结构,表明产物C有特殊的紫外吸收,我们将200 μL (缓冲液为200 mmol/L的硼酸硼砂缓冲液) 的底物和产物 (10 mmol-β-Phe、10 mmol丙酮酸、0.5 mmol 3-羰基-3-苯基丙酸) 分别在酶标仪上进行紫外扫描,检测产物C的特殊紫外吸收情况,来确定检测该反应的合适波长。确定好检测波长之后,测定底物及产物浓度和紫外吸收的关系,最终确定200 μL反应体系中,反应速率和紫外吸收之间的关系。
再次,在同样的反应体系中 (10 mmol底物,pH 8.2,浓度为100 mmol的硼酸硼砂缓冲液),加入纯酶3.5 μg/mL,分别用酶标仪和HPLC来测定反应的转化率情况,进行比较,来验证方法的可行性。
最后,方法确定后,用酶标仪检测不同底物浓度条件下的反应初速度。反应在200 mmol/L硼酸硼砂缓冲液 (pH 8.2)、室温25 ℃的条件下进行 (注:测定动力学常数时,使用的是第二批次纯化的酶,其他实验均为第一批次纯化的酶)。具体操作为:将1.89 μg的纯酶加入到96微孔板中,随后加入不同浓度的底物 (终体积为200 μL) 启动反应,用酶标仪检测该反应体系的紫外吸收变化,检测波长290 nm,时间为1 min,间隔为10 s,这样便可以测定不同底物浓度下的反应初速度。底物浓度的具体变化情况:当测定HBP对-β-Phe的动力学参数和底物抑制时,固定氨基受体丙酮酸的浓度为50 mmol/L,使-β-Phe的浓度为唯一变量,测定反应初速度;当测定氨基受体丙酮酸时,固定氨基供体-β-Phe的浓度为10 mmol/L,使丙酮酸的浓度为唯一变量。
酶活的定义:25 ℃下,在含有10 mmol/L-β-Phe和10 mmol/L丙酮酸的反应体系中,1 min内催化生成1 μmol产物 (丙氨酸或3- 羰基-3-苯基丙酸) 所需要的酶量,即1 U= 1 μmol/min。
1.5 底物谱的测定
HBP的氨基供体底物特异性采用典型的反应体系进行测定,但氨基供体更换成表2中的氨基化合物,浓度为10 mmol/L,丙酮酸的浓度为10 mmol/L,反应温度为40 ℃,pH 8.2 (100 mmol/L硼酸硼砂缓冲液)。反应5 min后,用HPLC检测产物丙氨酸的生成量来标定反应进行的程度,检测条件与1.4.1相同。丙氨酸的生成量越多,说明反应速度越快,酶活越高。其中,以-β-Phe为底物的反应为标准,将该反应丙氨酸的生成量设定为100%,其他氨基供体的活力采用相对活力来表示,即相对活力=其他氨基底物丙氨酸的生成量除以-β-Phe反应体系中丙氨酸的生成量×100%。测定氨基受体时,底物反之,条件亦然,反应结束后,用HPLC检测底物-β-Phe的减少量来标定反应进行的程度。
1.6 rac-β-Phe及其衍生物的手性拆分
采用典型的反应体系,氨基供体 (-β-Phe或其衍生物) 和丙酮酸的浓度均为10 mmol/L,反应温度为室温25 ℃,pH为8.2 (100 mmol/L硼酸硼砂缓冲液),加入66 μg的纯酶启动反应,反应12 h后,用HPLC检测产物的ee值和底物的转化率。ee值的检测条件与1.4.1条件一致;转化率的检测条件:色谱柱为CROWNPAK CR (+) column (4.0 mm×150 mm,5 μm),流动相为高氯酸水溶液 (加入10%甲醇,pH 1.5),流速为0.5 mL/min,等度洗脱,检测210 nm条件下的紫外吸收值。
2 结果与讨论
2.1 序列比对分析
HBP与已经报道的能够作用于芳香族β-氨基酸的ω-转氨酶 (分别来自sp. LUK、sp. JS666、和) 进行Pr-Blast,HBP与之同源性都较高,分别为51%、55%、59%和83%。这说明HBP极有可能作用于芳香族β-氨基酸。
2.2 HBP的表达纯化
将重组载体pET32a (+)-转化到BL21 (DE3) 中表达纯化,经过SDS-PAGE电泳分析,0.1 mmol IPTG、25 ℃诱导可以表达出大量的可溶性蛋白,经过镍柱纯化后即可除去大量杂质,得到纯酶;HBP的单亚基分子量约为45 kDa (图4),分子排阻实验测得该酶的分子量约为91.5 kDa,说明HBP由两个相同的亚基组成,与报道的大多数ω-转氨酶相同[25-26]。

图4 HBP的SDS-PAGE电泳检测
2.3 HBP的最适反应pH和最适反应温度
HBP的最适反应pH是通过测定其在不同的反应pH条件下的活力来确定的。观察实验结果,可以发现HBP喜碱厌酸,这一现象与已经报道的ω-转氨酶相似[27-28];HBP的最适反应pH在8.0–8.5之间 (图5a)。

图5 pH和温度对HBP活力的影响
选取pH 8.2的条件研究HBP的最适反应温度,结果表明,该酶的最适反应温度为40 ℃左右 (图5b),因此后续反应在此条件下进行。
2.4 HBP的pH稳定性和温度稳定性
HBP的稳定性是通过测定不同温度和pH条件处理后的残余活力来测定的。pH在6.0–10.0之间时,HBP的活性能够保持70%以上,而当pH降到酸性pH 5.0的时候,HBP的活力只有处理前的20%,表明酶的结构已经发生了严重变化,丧失了大部分活力 (图5c)。
由图5d可以看出温度对HBP稳定性的影响,在当温度低于50 ℃时,HBP的活力均能保持在80%以上,此时酶比较稳定;然而,当温度高于50 ℃后,活力出现大幅度下降,该酶不再稳定,温度到达60 ℃时,几乎丧失了全部活力。因此,在HBP的研究或应用过程中应该避免50 ℃以上的高温。
2.5 HBP的动力学参数和底物抑制情况
2.5.1 3-羰基-3-苯基丙酸的分解情况
β-酮酸是一种容易自发脱羧的不稳定化合物[24],因此我们研究了温度对产物C (3-羰基-3-苯基丙酸) 稳定性的影响。产物C的稳定性与温度呈负相关 (图6a),不同温度处理2 h后其分解产物苯乙酮随着温度的升高而增加;室温条件下,只有3%左右的酮酸产物C分解成苯乙酮,而当温度上升到60 ℃时,有65%左右的酮酸C分解成苯乙酮。产物C在25 ℃条件下的分解速率为0.03 %/min (图6b),分解速率极其缓慢,1 min内的分解对检测的影响可以忽略。实验初期,我们误以为该反应的主要推动力是产物C的减少;然而实验表明,产物C在室温25 ℃放置12 h后,大部分并没有分解 (大约14%分解),说明该反应平衡的主要推动力并不是产物的减少,而是该反应在此条件下的固有性质,即该反应的热力学平衡极其偏向于丙氨酸生成一方。这一结果,加深了我们对该反应历程的理解,对该反应的后续研究具有指导意义。
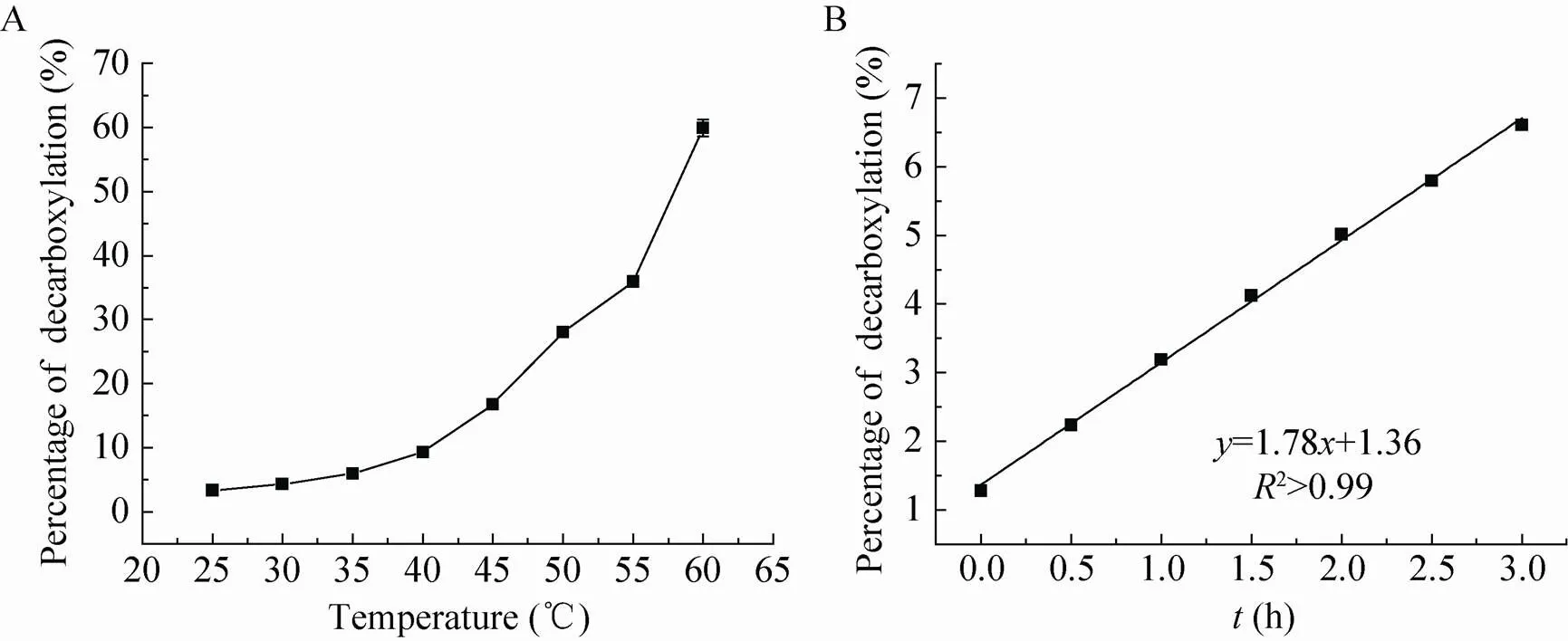
图6 温度对C分解的影响和分解速率
2.5.2 反应底物和产物的浓度与紫外吸收的关系
通过紫外全波长扫描,产物C在250 nm左右有最大的紫外吸收 (图7a),但是底物丙酮酸和β-Phe在250 nm下同样具有较强的紫外吸收;考虑到仪器的量程和背景吸收的问题,因此我们选择在290 nm的波长下进行检测。此条件下,底物和产物C的紫外吸收与浓度呈线性关系,符合朗伯比尔定律 (图7b、7c、7d)。由此我们可以计算出在200 μL的反应体系 (pH 8.2硼酸硼砂缓冲液) 下,底物浓度变化1.0 mmol/L,△290为0.73。

图7 底物和产物的全波长及其OD290与其浓度的关系
由此可知,酶的比活力计算公式为:
每分钟紫外吸收的变化(△290/△)×反应液总体积(L)/(0.73×酶量)。
其中,时间的单位为min,反应液总体积为常数200 μL,酶量的单位为mg。
2.5.3 紫外吸收法和HPLC方法的比较
相同条件下,酶标仪测定反应的转化率为5.97%±0.57%,HPLC测定反应的转化率为5.77%±0.44%。两种方法测量的结果差距较小,因此可以采用紫外吸收法测定该反应的反应速率。并且计算得到酶活为33.80 U/mg。
2.5.4 底物浓度对酶活的影响
采用以上建立的紫外吸收法,测定不同底物浓度条件下反应的酶活,这样不仅可以计算出HBP的动力学参数,还可以了解底物的抑制情况。根据不同底物浓度下的反应初速度,由sigmaplot软件计算出HBP对-β-Phe和丙酮酸的m分别是1.6 mmol/L和8.6 mmol/L,cat分别为142.3 s–1和148.5 s–1,此时max分别为 88.5 μmol/min·mg和92.9 μmol/min·mg (图8)。底物浓度升高时,显示出微弱的底物抑制现象,当-β-Phe的浓度增加到80 mmol时,其活力为83.28 μmol/min·mg;当丙酮酸的浓度升高到 200 mmol时,其反应速率为70.98 μmol/min·mg。

图8 HBP对底物的米氏方程曲线
2.6 HBP的底物谱情况
2.6.1 氨基供体底物谱
HBP的氨基供体特性是以丙酮酸为氨基受体来测定的。由表1可以看出HBP对不同种类的芳香族β-氨基酸都有活性,并且活力差别不大,比较A5和A6,这两种底物的侧链都是给电子基团,A5的活力低于A1,A6的活力高于A1,因此,芳香族β-氨基酸苯环侧链的不同,对位基团的供给电子能力并不能对反应活性造成绝对的影响,反应活力的高低取决于底物和酶的综合性相互作用。比较特别的是,HBP对脂肪族β-氨基酸A8没有活性,却对含有支链的脂肪族β-氨基酸A7活力较高,表明HBP的小口袋比较大,只能接受侧链相对较大的基团,不能接受侧链比较小的基团。有意思的是,对于典型的氨基供体A9 (苯乙胺),几乎所有报道的ω-转氨酶都对其表现出高活性[5,11],但是HBP对其却没有任何活性。由上可知,HBP催化的底物比较专一,对芳香族β-氨基酸和支链的脂肪族β-氨基酸有特异性。
表1 HBP的氨基供体底物谱
Table 1 Amino donors specificity of HBP

2.6.2 氨基受体底物谱
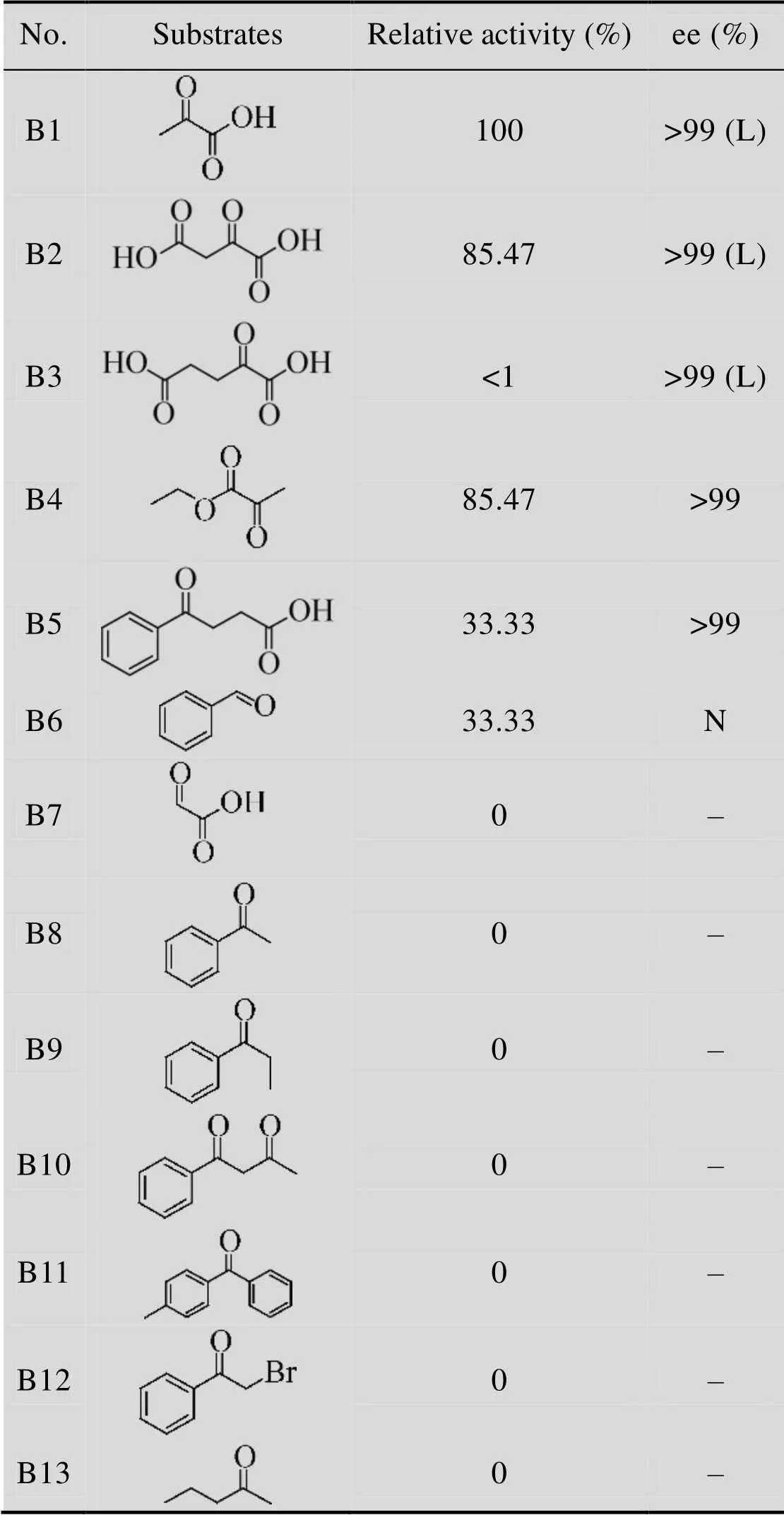
HBP的氨基受体底物特性是以-β-Phe为氨基供体来测定的。结果显示,与其他能够作用于芳香族β-氨基酸的转氨酶[20-22,25]相似,HBP能够作用于含有羧酸基团的底物B1、B2和B3,并且表现出完美的立体选择性,相对应的产物ee值在99%以上。对于B4,HBP表现出较高的活力,有意思的是,检测到B4对应的产物为L-Ala,由于检测条件是碱性的,底物B4和其对应的产物都是酯类,所以无法确定HBP能否以丙酮酸乙酯为氨基受体;可以推测,此反应存在两种可能:第一,HBP以B4为底物,反应生成手性的丙氨酸乙酯在碱性条件下水解生成L-Ala;第二,HBP不能以B4为底物进行反应,而是底物B4在碱性条件下先水解成丙酮酸,然后HBP以丙酮酸为底物进行反应。对于B5,其对应的产物是具有应用价值的γ-氨基酸[29],可惜的是当HBP以-β-Phe作为氨基供体时,得到的两种产物 (3-氨基-3-苯基丙酸和4-氨基- 4-苯基丁酸) 结构上非常相似从而很难分离;对于醛类B6是一种廉价从而非常适合进行拆分的氨基受体。有趣的是,HBP对B7没有活性,B7与B1相比,拥有更小的位阻,理论上应该具有相似的活性,但是HBP对B7没有活力,我们推测其底物结合口袋处,有一些特殊的结构而导致这一现象,希望之后能够通过三维结构得到解释;其他底物,B8到B13,其中有一些和标准底物 (β-Phe和丙酮酸) 的结构很相近,但是HBP对这些底物都没有催化活性。总之,HBP催化的底物相对比较专一,只对特有的底物有活力。
表2 HBP的氨基受体底物谱
Table 2 Amino acceptors specificity of HBP
2.7 rac-β-Phe及其衍生物的手性拆分
-β-Phe及其衍生物的手性拆分是在典型的反应体系中测定的。首先我们测定HBP对 10 mmol/L的-β-Phe及其衍生物的拆分情况。反应12 h后,检测结果如表3所示,结果显示,对于-β-Phe及其不同的衍生物,HBP均能有效地将其进行手性拆分,最终得到光学纯的芳香族β-氨基酸产物。其转化率均在50%左右,ee>99%。
表3 HBP动力学拆分-β-Phe及其衍生物
Table 3 Preparation of ()-Phe and its derivatives using HBP-catalyzed kinetic resolution of racemic substrates

为了进一步研究HBP手性拆分芳香族β-氨基酸的能力,选取-β-Phe为代表,逐步提高底物的浓度;采用典型的反应体系,但有一些不同,缓冲液采用200 mmol/L的硼酸硼砂缓冲液 (通过增加缓冲液的浓度来增加缓冲液的缓冲能力,pH 8.2)。底物-β-Phe的浓度增加到100 mmol/L (接近饱和) 时,氨基受体采用 100 mmol/L的丙酮酸钠 (丙酮酸钠取代丙酮酸,防止pH变化过大);反应进行7 h,β-Phe的ee值大于99%,说明HBP可以将100 mmol/L的-β-Phe有效地拆分 (图9)。当我们继续增加底物浓度,到150 mmol/L时,反应初期底物-β-Phe处于溶解平衡状态,随着反应的进行,-β-Phe逐渐溶解,反应体系逐渐清澈,24 h后补加66 μg的纯酶,总反应进行36 h时,ee值达到97.47%。

图9 HBP动力学拆分100 mmol/L的rac-β-Phe
2.8 几种不同ω-转氨酶的性质比较
根据前面的实验结果,我们将HBP与最近报道的能够作用于芳香族β-Phe的ω-转氨酶进行了简单的比较。通过比较发现,HBP的最适反应温度与其他几个酶比较相似,最适反应pH都偏向于碱性条件。手性拆分的底物浓度比较高 (150 mmol),不足之处在于不能进行β-Phe的不对称合成 (表4)。
表4 几种不同ω-转氨酶的性质比较
Table 4 Comparison of different ω-transaminases

3 结论
本研究中,采用基因挖矿的方法从 NCBI数据库中发现了一个来自PsJN的新型 () 立体选择性ω-转氨酶,并将其在BL21 (DE3) 中进行异源表达,之后测定了HBP的酶学性质、动力学参数并研究了其底物谱。HBP最适反应pH在8.0–8.5之间,最适反应温度为40 ℃。测定过程中,建立了一种简便快捷的检测方法,加深了我们对β-Phe脱氨反应热力学平衡的认识。此酶表现出严格的 () 立体选择性和相对专一的底物选择性。对于氨基供体,HBP能特异性地识别侧链比较大的芳香族β-氨基酸,包括芳香族β-氨基酸和带有支链的脂肪族β-氨基酸,与已经报道的ω-转氨酶不同的是,HBP不能以苯乙胺为氨基供体。对于氨基受体,该酶对比较常见的氨基受体丙酮酸的活力较高,能够以B5 (4-羰基-4-苯基丁酸) 为氨基受体,不对称合成芳香族γ-氨基酸,遗憾的是我们尚未发现理想的氨基供体。总体而言,HBP是一种可以有效地拆分芳香族β-氨基酸的生物催化剂。
REFERENCES
[1] Zhu DM, Hua L. Biocatalytic asymmetric amination of carbonyl functional groups-a synthetic biology approach to organic chemistry. Biotechnol J, 2009, 4(10): 1420–1431.
[2] Koszelewski D, Tauber K, Faber K, et al. ω-Transaminases for the synthesis of non-racemic α-chiral primary amines. Trends Biotechnol, 2010, 28(6): 324–332.
[3] Bessman SP, Rossen J, Layne EC. γ-Aminobutyric acid-glutamic acid transamination in brain. J Biol Chem, 1953, 201(1): 385–391.
[4] Stirling DI, Zeitlin AL, Matcham GW. Enantiomeric enrichment and stereoselective synthesis of chiral amines: US, 4950606. 1990-08-21.
[5] Mathew S, Yun H. ω-Transaminases for the production of optically pure amines and unnatural amino acids. ACS Catal, 2012, 2(6): 993–1001.
[6] Malik MS, Park ES, Shin JS. Features and technical applications of ω-transaminases. Appl Microbiol Biotechnol, 2012, 94(5): 1163–1171.
[7] Höhne M, Bornscheuer UT. Application of transaminases//Drauz K, Gröger H, May O, Eds. Enzyme Catalysis in Organic Synthesis. 3rd ed. Weinheim: Wiley-VCH, 2012: 779–820.
[8] Matcham G, Bhatia M, Lang W, et al. Enzyme and reaction engineering in biocatalysis: synthesis of ()-methoxyisopropylamine(=()-1-Methoxypropan-2-amine). CHIMIA, 1999, 53(12): 584–589.
[9] Savile CK, Janey JM, Mundorff EC, et al. Biocatalytic asymmetric synthesis of chiral amines from ketones applied to sitagliptin manufacture. Science, 2010, 329(5989): 305–309.
[10] Jiang JJ, Chen X, Feng JH, et al. Substrate profile of an ω-transaminase fromand its potential for the production of optically pure amines and unnatural amino acids. J Mol Catal B: Enzym, 2014, 100: 32–39.
[11] Shin JS, Kim BG. Exploring the active site of amine: pyruvate aminotransferase on the basis of the substrate structure-reactivity relationship. J Org Chem, 2002, 67(9): 2848–2853.
[12] Nobili A, Steffen-Munsberg F, Kohls H, et al. Engineering the active site of the amine transaminase fromfor the asymmetric synthesis of aryl-alkyl amines and amino alcohols. ChemCatChem, 2015, 7(5): 757–760.
[13] Höhne M, Schätzle S, Jochens H, et al. Rational assignment of key motifs for function guidesenzyme identification. Nat Chem Biol, 2010, 6(11): 807–813.
[14] Jiang JJ, Chen X, Zhang DL, et al. Characterization of ()-selective amine transaminases identified bymotif sequence blast. Appl Microbiol Biotechnol, 2015, 99(6): 2613–2621.
[15] Grayson JI, Roos J, Osswald S. Development of a commercial process for ()-β-phenylalanine. Org Process Res Dev, 2011, 15(5): 1201–1206.
[16] Preiml M, Hillmayer K, Klempier N. A new approach to β-amino acids: biotransformation of-protected β-amino nitriles. Tetrahedron Lett, 2003, 44(27): 5057–5059.
[17] Turner NJ. Ammonia lyases and aminomutases as biocatalysts for the synthesis of α-amino and β-amino acids. Curr Opin Chem Biol, 2011, 15(2): 234–240.
[18] Zhang DL, Chen X, Zhang R, et al. Development of β-amino acid dehydrogenase for the synthesis of β-amino acidsreductive amination of β-keto acids. ACS Catal, 2015, 5(4): 2220–2224.
[19] Rudat J, Brucher BR, Syldatk C. Transaminases for the synthesis of enantiopure β-amino acids. AMB Express, 2012, 2(1): 11.
[20] Kim J, Kyung D, Yun H, et al. Cloning and characterization of a novel β-transaminase fromsp. strain LUK: a new biocatalyst for the synthesis of enantiomerically pure β-amino acids. Appl Environ Microbiol, 2007, 73(6): 1772–1782.
[21] Bea HS, Park HJ, Lee SH, et al. Kinetic resolution of aromatic β-amino acids by ω-transaminase. Chem Commun, 2011, 47(20): 5894–5896.
[22] Crismaru CG, Wybenga GG, Szymanski W, et al. Biochemical properties and crystal structure of a β-phenylalanine aminotransferase from. Appl Environ Microbiol, 2013, 79(1): 185–195.
[23] Mathew S, Bea H, Nadarajan SP, et al. Production of chiral β-amino acids using ω-transaminase from. J Biotechnol, 2015, 196–197: 1–8.
[24] Bach RD, Canepa C. Electronic factors influencing the decarboxylation of β-keto acids. A model enzyme study. J Org Chem, 1996, 61(18): 6346–6353.
[25] Wybenga GG, Crismaru CG, Janssen DB, et al. Structural determinants of the β-selectivity of a bacterial aminotransferase. J Biol Chem, 2012, 287(34): 28495–28502.
[26] Rausch C, Lerchner A, Schiefner A, et al. Crystal structure of the ω-aminotransferase fromand its phylogenetic relationship with other class III aminotransferases that have biotechnological potential. Proteins, 2013, 81(5): 774–787.
[27] Hwang BY, Ko SH, Park HY, et al. Identification of ω-Aminotransferase fromand sitedirected mutagenesis to broaden substrate specificity. J Microbiol Biotechnol, 2008, 18(1): 48–54.
[28] Kaulmann U, Smithies K, Smith MEB, et al. Substrate spectrum of ω-transaminase fromDSM30191 and its potential for biocatalysis. Enzyme Microb Technol, 2007, 41(5): 628–637.
[29] Shon M, Shanmugavel R, Shin G, et al. Enzymatic synthesis of chiral γ-amino acids using ω-transaminase. Chem Commun, 2014, 50(84): 12680–12683.
Expression and characterization of a novel ω-transaminase fromPsJN
Yuncheng Du1,2, Wenyue Dong1, Jinju Jiang1,2, Qijia Chen1,2, Jinhui Feng1, Qiaqing Wu1, and Dunming Zhu1
1 National Engineering Laboratory for Industrial Enzymes, Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences, Tianjin 300308, China 2 College of Life Science, University of Chinese academy of science, Beijing 100049, China
Production of chiral amines and unnatural amino-acid using ω-transaminase can be achieved by kinetic resolution and asymmetric synthesis, thus ω-transaminase is of great importance in the synthesis of pharmaceutical intermediates. By genomic data mining, a putative ω-transaminase genewas found inPsJN. The genewas cloned and over-expressed inBL21 (DE3). The recombinant enzyme (HBP) was purified by Ni-NTA column and its catalytic properties and substrate profile were studied. HBP showed high relative activity (33.80 U/mg) and enantioselectivity toward β-phenylalanine (β-Phe). The optimal reaction temperature and pH were 40 ℃ and 8.0–8.5, respectively. We also established a simpler and more effective method to detect the deamination reaction of β-Phe by UV absorption method using microplate reader, and demonstrated the thermodynamic property of this reaction. The substrate profiling showed that HBP was specific to β-Phe and its derivatives as the amino donor. HBP catalyzed the resolution of-β-Phe and its derivatives, the products ()amino acids were obtained with about 50% conversions and 99%.
ω-transaminase,PsJN, aromatic β-amino acids, chiral resolution
October 27, 2015; Accepted: February 17, 2016
工业生物技术
杜允成, 董文玥, 姜进举, 等. 一种来源于PsJN的ω-转氨酶的表达纯化及性质分析. 生物工程学报, 2016, 32(7): 912–926.
Du YC, Dong WY, Jiang JJ, et al. Expression and characterization of a novel ω-transaminase fromPsJN. Chin J Biotech, 2016, 32(7): 912–926.
Supported by: National Basic Research Program of China (973 Program) (No. 2011CB710800).
Corresponding authors: Qiaqing Wu. Tel: +86-22-84861963; Fax: +86-22-84861996; E-mail: wu_qq@tib.cas.cn
Dunming Zhu. Tel: +86-22-84861962; Fax: +86-22-84861996; E-mail: zhu_dm@tib.cas.cn
国家重点基础研究发展计划 (973计划) (No. 2011CB710800) 资助。
(本文责编 郝丽芳)
