还原条件对甲烷催化裂解催化剂活性影响探究
2016-04-12梁威陈晨李腾陈小博山红红
梁威,陈晨,李腾,陈小博,山红红
(中国石油大学(华东)化学工程学院,重质油国家重点实验室,山东青岛266580)
还原条件对甲烷催化裂解催化剂活性影响探究
梁威,陈晨,李腾,陈小博,山红红
(中国石油大学(华东)化学工程学院,重质油国家重点实验室,山东青岛266580)
用溶胶凝胶法制备了w(Ni)=50%的Ni/Al2O3催化剂,考察了不同还原条件对其催化裂解甲烷性能的影响。对新鲜的和使用后的催化剂进行了N2物理吸附、XRD和H2-TPR表征。结果表明:随着催化剂还原时间的延长,甲烷转化率先增加后降低,在还原90min时达最高,而氢气选择性则是逐渐增高;随着催化剂还原温度的提高,甲烷转化率呈现先增加后降低的趋势,氢气选择性则呈现逐渐增加的趋势,在800℃以上趋于100%。表征结果表明焙烧后的催化剂上存在3种不同形式的NiO,在还原过程中,自由态的NiO最易被还原,分散态的NiO次之,固定态的NiO最难被还原。将催化剂活性的降低归结于催化剂上大晶粒的镍物种在高温H2氛围下发生的烧结现象。还原时间的延长有利于催化剂中NiO物种被还原,但是同时也发现,单纯通过延长还原时间无法达到完全还原催化剂中镍物种的目的。随着还原温度的提高,催化剂中的镍物种被完全还原,然而催化剂的烧结现象严重。因此,选择合适的还原条件将有利于催化剂表现出较高的活性。
甲烷;催化裂解;氢气;催化剂;还原时间;还原温度
氢气广泛用于化工、炼油、电子、玻璃生产等众多行业,市场需求量巨大,目前我国在合成氨、石油化工和甲醇生产等领域每年需氢气超过2000亿m3。煤和石油等化石燃料的大量非清洁使用使得环境不断恶化,开发能源利用率高的环境友好型能源成为各国积极研究的方向。氢气因来源广泛、热值高和燃烧产物无污染,是理想的替代传统化石燃料的新能源,因此氢能开发成为研究的热点[1-3]。因此,开发清洁廉价的氢气制备方法便显得尤为重要。氢气的制备方法多种多样,主要有天然气制氢,电解水,煤气化和生物质制氢等[4-9],目前工业上国外主要采用天然气制氢,我国则是煤制氢占主导。然而,在这两种制氢过程中,均会产生大量的温室气体CO2,且工艺流程长,投资大,能耗和成本高[10]。甲烷催化裂解制氢由于其较低的能耗(ΔH=-75.6kJ/mol),在反应过程中气相产物以H2为主,只有极少量的COx生成,并且可以副产碳纳米管等高附加值的产品等优点,正逐渐受到人们的关注。
在甲烷催化裂解制氢反应催化剂的研究当中,Ni/Al2O3研究较为广泛[11-13],金属镍由于其良好的脱氢活性和低廉的价格,成为活性金属的较好选择,Al2O3作为载体具有较高的高温稳定性和较大的比表面积等优点,同时,Ni与Al2O3之间存在较强的相互作用,可以有效提高活性金属在载体上的分散度并抑制催化剂在再生过程中的烧结现象发生。对于以镍为活性金属的催化剂,在高温焙烧后,镍将以NiO的形式存在于载体的表面,而催化甲烷裂解的活性位为Ni0,因此在催化剂使用前需要进行H2预还原处理,将NiO还原为Ni0。高温以及H2的存在将对催化剂的活性和组织结构产生较大影响,而文献对H2预还原对催化剂的反应活性影响的报道较少,因此,本文将从还原时间和还原温度两个方面对H2预还原对催化剂活性的影响进行探究。
1 实验部分
1.1 催化剂的制备
实验选用的w(Ni)=50%的Ni/Al2O3催化剂采用溶胶凝胶法制备,具体步骤如下:称取一定质量的拟薄水铝石(中国铝业股份有限公司山东分公司生产)。加入适量的去离子水,搅拌均匀。搅拌的同时逐滴加入一定质量的浓盐酸,充分搅拌后形成氧化铝凝胶。将该氧化铝凝胶置于65℃水浴中加热,调节溶液pH值3~4,搅拌30min后取出,向溶液中加入计量量的Ni(NO3)2·6H2O(国药集团化学试剂有限公司生产),继续在室温下搅拌1.5h。将搅拌好的凝胶放入烘箱中烘干4~6h。最后将烘干的催化剂样品置于马弗炉中700℃焙烧2h,升温速率10℃/min。将焙烧后的催化剂研磨,筛取粒度为80~180目的催化剂待用。
1.2 催化剂的表征
催化剂的物相结构采用荷兰Panalytical公司生产的X’Pert PRO MPD衍射仪测定,其光源为Cu靶Kα辐射,管电压40kV,管电流40mA,扫描步长0.0167°,在2θ为10~80°的范围内记录衍射谱图。
低温N2吸附-脱附实验在Quanta公司生产的Quadrasorb SI物理吸附仪上进行,样品比表面积由BET计算方法得到,孔体积及孔径分布依据BJH方法计算得到。
H2-TPR分析在浙江泛泰仪器生产的FINESORB-3010型程序升温化学吸附仪上进行,采用φ(H2)为10%的氢氮混合气作为气体吸附质,气体流速30mL/min,采用TCD检测器进行检测,TCD电流30mA,测温区间80~1000℃。
1.3 催化剂的性能评价
在固定床微型反应器上对催化剂催化甲烷裂解制氢性能进行评价。整个反应阶段分为H2预还原阶段和催化CH4裂解反应两个阶段。反应器材质为石英,内径6mm,催化剂装填量0.5g,首先在30mL/min纯度为99.9%的N2保护下以10℃/min的升温速率升温至预定还原温度,然后切换为20mL/min纯度为99.999%的H2进行预定时间的预还原,还原结束后切换为30mL/min的N2吹扫30min。待吹扫结束,在700℃下进行甲烷裂解反应,CH4流量为30mL/min,纯度99.999%,进料时间2min,采用排水集气的方法收集气体并计算体积。气体产物采用北京北分瑞利分析仪器公司生产的SP-3420A气相色谱进行分析。
2 结果与讨论
2.1 还原时间对催化剂甲烷裂解性能的影响
2.1.1 不同时间H2还原后的催化剂BET表征
在700℃下,对催化剂分别进行30min、60min、90min、120min、150min、180min的H2预还原,对未经还原和经过还原后的催化剂进行低温N2吸脱附实验。由图1可以看出,没有经过还原的催化剂和经过不同时间还原的催化剂均呈现了IV型等温曲线,均存在明显的滞后回环,说明未经还原的催化剂和经过还原后的催化剂都形成了良好的介孔结构。
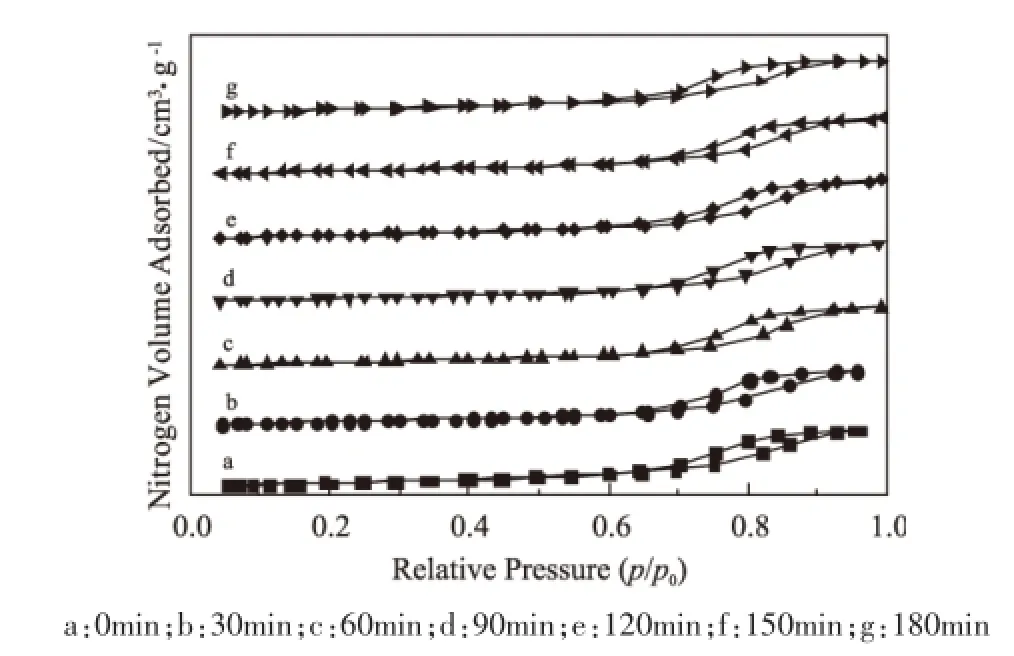
图1 不同时间H2还原后的催化剂的N2吸附-脱附等温线
图2是经过不同还原时间后的催化剂的孔径分布图,由图可看出,未经还原的催化剂孔径主要分布在7.8nm左右,而对于经过还原后的催化剂,随着H2还原时间的延长,7.8nm左右的孔逐渐减少,9.5nm左右的孔逐渐增多,这可能与催化剂中物相的转变有关。
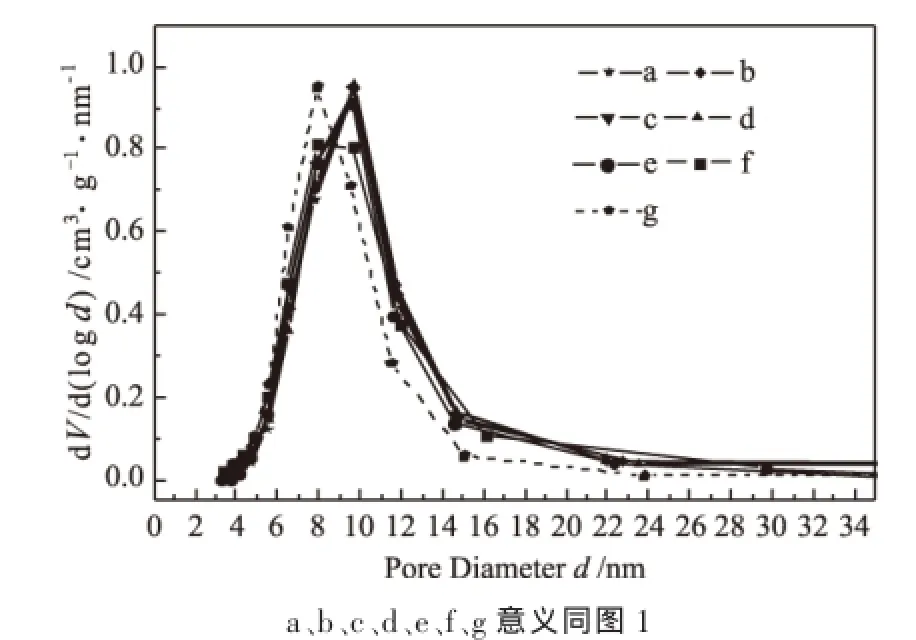
图2 不同时间H2还原后催化剂的孔径分布
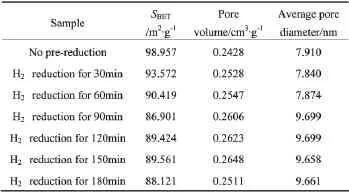
表1 经过不同预还原时间后催化剂的比表面积和孔结构性质
表1为经过不同时间H2还原后催化剂的比表面积和孔结构性质,由表中可以看出,随着还原时间的延长,催化剂的比表面积逐渐减少,这是由于H2对催化剂组织结构的破坏引起的。同时,在NiO被还原成Ni0的过程中,会发生镍晶粒的烧结,同样会导致比表面积的降低。从表1中还可看出,经过还原的催化剂与未经还原的催化剂相比,未发生较大的孔容变化。
2.2.2 不同时间H2还原后的催化剂XRD表征
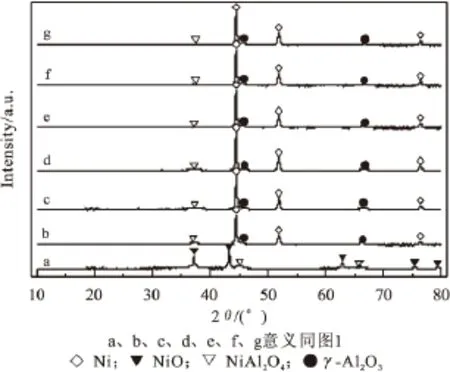
图3 经过不同还原时间后的催化剂XRD图
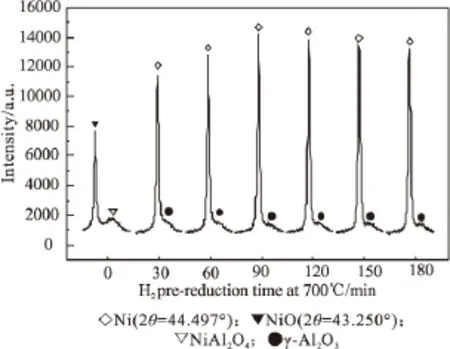
图4 不同还原时间后的催化剂XRD图(2θ=42~47°)
分别对未经过还原的催化剂和经过不同还原时间后的催化剂进行了XRD物相表征。由图3可以看出,在未经过还原的催化剂的XRD图中,可以看到明显的NiO物相(2θ=43.250°、37.207°、62.787°、62.861°、75.294°),以及NiAl2O4物相(2θ= 65.495°、36.963°、45.068°),说明除形成尖晶石结构外,还存在大量自由态的NiO。经过不同时间的H2还原之后,XRD图中均可以看到明显的Ni晶相(2θ=44.497°、51.851°、76.383°),另外还可以看到明显的γ-Al2O3物相(2θ=45.915°、66.951°),以及少量的NiAl2O4物相(2θ=36.963°),值得注意的是,在经过了120min和180min的H2还原后的催化剂XRD图中,都仍可看到NiAl2O4的物相存在,因此,在700℃的还原温度下,通过延长还原时间,无法达到完全还原催化剂中镍物种的目的。图4选取了经过不同H2还原时间的催化剂中Ni在2θ=42°~47°的XRD谱图进行对比,发现随着还原时间的延长,Ni的特征还原峰逐渐增强,在经过预还原90min后,Ni的特征还原峰达到最强,继续延长还原时间,Ni的特征衍射峰逐渐降低。推测,在经过90min还原后,还原出的单质镍物种最多,随着还原时间的继续延长,被还原出的单质镍物种在H2氛围下出现烧结现象,还原时间越长,烧结现象越严重。
2.2.3 不同时间H2还原后的催化剂TPR表征
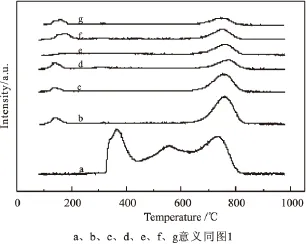
图5 不同预还原时间的催化剂H2-TPR图
对未经过还原的催化剂和经过不同时间H2还原后的催化剂进行了H2-TPR表征,由图5可以看出,未经过还原的催化剂出现3个H2吸收峰。还原峰温分别在320~440℃、440~640℃、640~820℃。许多研究者的研究已经表明,在NiO/Al2O3体系当中,NiO将以多种不同的形式存在于Al2O3上,Zielineski等[14]认为在γ-Al2O3的表面存在着两种状态的NiO,一种为保持着体相NiO性质的自由态的NiO,另一种为固定态的NiO,这种固定态的形成可能是由于NiO与γ-Al2O3载体表面形成以表面态计量或非计量的铝酸盐。Scheffer等[15]通过研究将Ni的存在形式分为以下3种,并对其还原峰温进行了归属:430~630℃为高分散无定型表面Ni2+的还原,他们认为这是一种八面体配位的Ni,770℃左右的还原峰归属为一种表面NiO,870℃左右的还原峰认为是一种同时存在于表面和载体中的NiAl2O4的还原。张玉红等[16]通过XRD和TPR的结果,将Ni在Al2O3上的存在形式归为3类:自由态的NiO、分散态的NiO和固定态的NiO,而又将固定态的NiO细分为微晶态的NiAl2O4、晶相NiAl2O4和类尖晶石结构的固溶体等3种。同时他们还指出,自由态的NiO是由大晶粒的NiO聚集而成,在H2还原的过程中,将形成比表面积小的大晶粒尺寸的Ni0,这种Ni0的催化活性很低,对于分散态的NiO和固定态的NiO,都可以经过还原形成小晶粒的Ni0,与大晶粒的Ni0相比,增加了反应的活性位,使反应活性得到提高,但是这种被还原出来的Ni0与载体之间的相互作用较弱,容易发生烧结。而对于固定态的NiO,在还原气氛下还原出的Ni0周围有许多Al2O3,并且有很强的相互作用,从而能够抑制Ni0的流失与烧结。
结合文献报道,我们将这未经过还原的催化剂的TPR图中的3种还原峰归属如下:320~440℃的还原峰归属于在Al2O3表面与载体相互作用较弱的自由态NiO,这种NiO物种容易发生团聚,以大晶粒的形式存在;440~640℃的还原峰归属为与载体有一定相互作用的分散态的NiO,这种形式的NiO以Ni-O-Al的键合形式存在;640~820℃的还原峰归属为固定态的NiO晶相,Ni以NiAl2O4的尖晶石相存在。
2.2.4 不同时间H2预还原后的催化剂活性评价
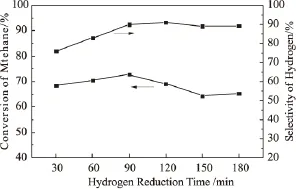
图6 不同预还原时间对催化剂活性的影响
对经过不同时间H2预还原后的催化剂在实验室固定床微反装置上进行甲烷催化裂解实验。由图6可以看出,随着催化剂还原时间的延长,CH4转化率呈现先增加后降低的趋势,H2选择性则呈现先增加后保持基本不变。在考察的还原时间内,H2还原时间为30min时,从图5中可以看出,归属于自由态和分散态的NiO的H2特征还原峰消失,与未经还原的催化剂相比,归属于固定态NiO的H2特征还原峰变小,说明已经不存在自由态的NiO和分散态的NiO,而且已有部分固定态的NiO(NiAl2O4)得到了还原,随着还原时间的延长,有更多的固定态的NiO,即存在于NiAl2O4中的Ni被还原,被还原出的作为脱氢活性位的Ni0增多,因此甲烷的转化率逐渐提高,同时在还原的过程中,催化剂的表面氧物种和晶格氧不断被消耗,减少了生成的H2与氧物种的反应,因此使得H2选择性不断提高。然而,当还原时间大于90min后继续延长还原时间,CH4转化率和H2选择性逐渐下降,推测这是由较易烧结的自由态和分散态的NiO在H2气氛下发生烧结引起的,由图4可以看出,在2θ=44.497°的Ni的特征衍射峰逐渐降低,我们将这归因于还原出的Ni0发生了烧结团聚。同时也注意到,当预还原时间在90min后继续延长时,由图5还原后的TPR图可以看出,催化剂中仍然存在一定量的固定态的NiO(NiAl2O4)无法被还原,因此,单纯通过延长还原时间的方法,无法达到提高选择性的目的。
2.3 不同H2预还原温度对催化剂反应活性的影响
2.3.1 不同温度H2还原后的催化剂BET表征
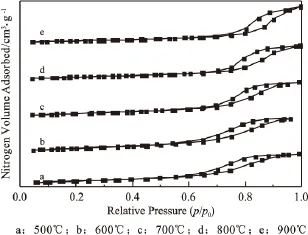
图7 不同温度H2预还原后的催化剂N2吸附-脱附等温线
通过前期对H2预还原时间的考察,确定90min为最优还原时间,在此基础上考察不同预还原温度对催化剂活性的影响,结果如图7。通过图7可以看出,经过不同温度还原后,催化剂仍然表现出IV型吸附峰温线,存在明显的滞后回环,因此具有较好的介孔结构,但从表2可以看出,预还原温度的提高导致催化剂的比表面积逐渐减小,平均孔径和孔容都呈现逐渐增大的趋势,这说明高温下Ni的存在使催化剂的部分孔道发生坍塌,高温对催化剂的组织结构破坏严重。
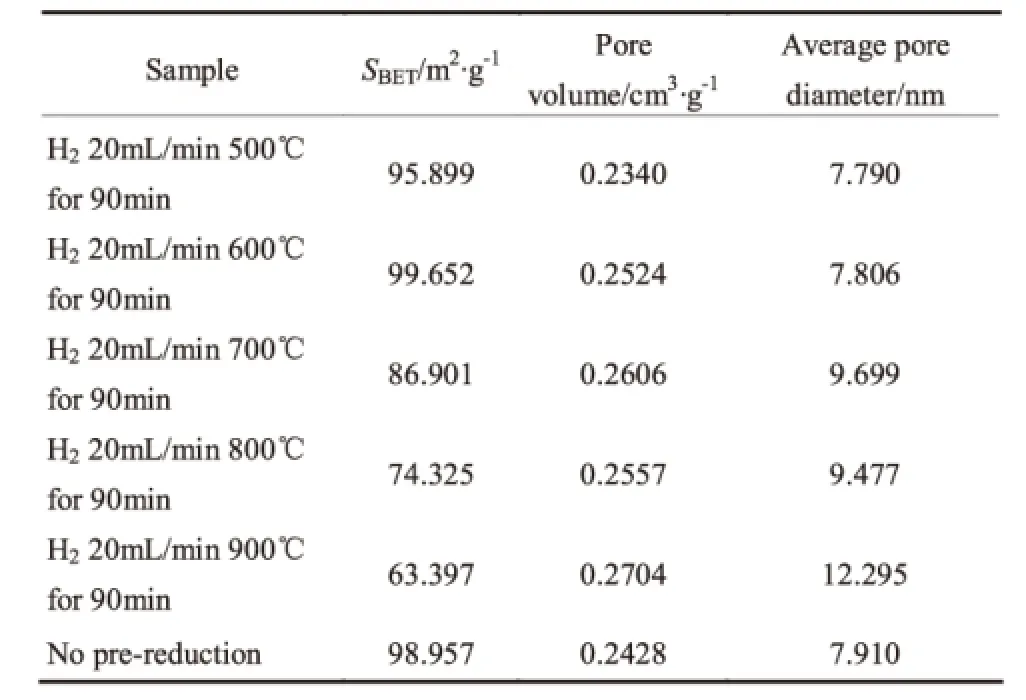
表2 不同温度H2还原后催化剂的比表面积和孔结构性质
2.3.2 不同温度H2还原后的催化剂XRD表征
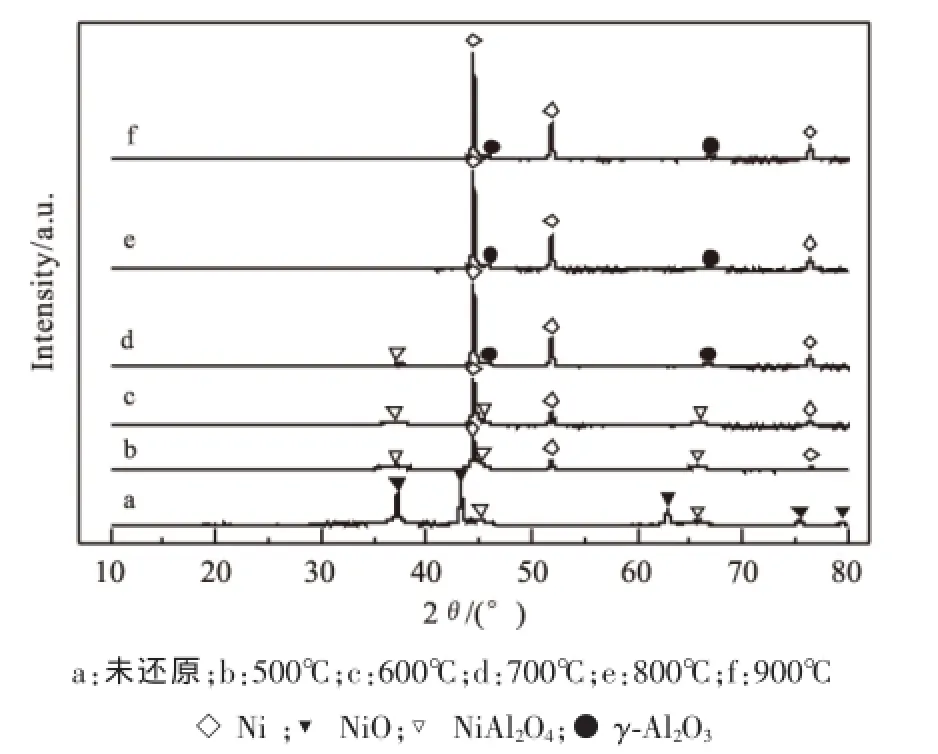
图8 不同温度H2预还原后的催化剂XRD图
图8为经不同温度H2预还原后的催化剂的XRD表征结果。由图8可看出,500℃和600℃后的催化剂上主要表现出Ni(2θ=44.497°、51.851°、76.383°)、NiAl2O4(2θ=36.963°、45.068°、65.495°)物相,在700℃预还原后除表现出Ni、NiAl2O4的特征衍射峰外,还出现了γ-Al2O3(2θ=45.912°、66.951°)的特征衍射峰。经过800℃、900℃预还原之后,表现出Ni、γ-Al2O3的特征衍射峰,而很难观察到NiAl2O4的特征衍射峰。通过XRD表征可以发现,较高的还原温度有利于将催化剂中的镍物种完全还原。
2.3.3 不同温度H2还原后的催化剂TPR表征
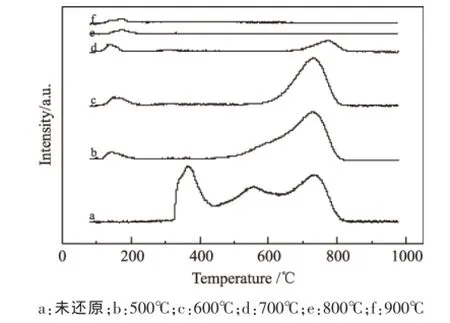
图9 不同温度H2预还原后的催化剂H2-TPR图
图9为不同温度下H2还原后的催化剂H2-TPR表征结果。由图9可看出,经过500℃还原后的催化剂在450~820℃之间有2个H2还原峰,说明仍然存在2种不同形式的NiO,一种为还原峰温为440~640℃的分散态的NiO,另一种为与载体有较强相互作用的NiO,即存在于NiAl2O4中的镍物种。经过600℃和700℃预还原后的催化剂的H2-TPR图中仅可以看到还原峰温大于640℃的还原峰,因此未被还原的镍物种仅存在于NiAl2O4中。另外可以看出,经过700℃还原后的催化剂的H2-TPR图中的高温还原峰面积逐渐降低,且峰温向高温处移动。张玉红等[16]又将固定态的NiO细分为微晶态的NiAl2O4、晶相NiAl2O4和类尖晶石结构的固溶体等3种,因此,我们将峰温向高温处移动归因于还原峰温在640~820℃的固定态的NiO中还存在不同形式的NiO,它们的还原难易程度不同。在经过800℃和900℃还原后的催化剂的H2-TPR图中,已经观察不到H2的消耗峰,说明催化剂中的NiO已经被完全还原为Ni0。综合H2-TPR的表征结果,还可以得出结论,催化剂中NiO物种被还原的难易程度为自由态的NiO最易被还原,分散态的NiO次之,固定态的NiO最难被还原。
2.3.4 不同温度H2预还原后的催化剂的活性评价
图10为不同温度H2预还原后的催化剂甲烷裂解活性评价结果。由图10可看出,在还原时间为90min时,随着还原温度的提高,CH4转化率呈现先增加后降低的趋势,H2选择性则逐渐增加并向100%趋近。这是因为,还原温度的提高有利于催化剂中NiO的还原,还原成的作为脱氢活性位的Ni0逐渐增多,载体上的表面氧和晶格氧逐渐减少,因此CH4转化率和H2选择性逐渐增加,当还原温度高于700℃后继续提高还原温度,将加速已经还原出的自由态和分散态的镍物种的烧结团聚,进而生成比表面积小的大晶粒镍物种,使得催化剂的活性降低。
3 结论
选取溶胶凝胶法制备的w(Ni)=50%的Ni/Al2O3催化剂,考察不同还原条件对其催化裂解甲烷活性的影响。通过实验发现,随着还原时间的延长,催化剂的甲烷转化率先增加后降低,氢气选择性逐渐增高,通过表征发现,还原时间的延长有利于催化剂中NiO物种被还原,但是同时也发现,单纯通过延长还原时间无法达到完全还原催化剂中镍物种的目的。其次,考察了还原温度对反应活性的影响,通过实验发现,随着还原温度的提高,催化剂的甲烷转化率呈现先增加后降低的趋势,氢气选择性呈现逐渐增加的趋势,在温度高于800℃时趋于100%。通过表征发现,随着还原温度的提高,催化剂的烧结现象严重。因此,选择合适的还原条件将有利于催化剂表现出较高的活性。
[1]Conte M,Iacobazzi A,Ronchetti M,et al.Hydrogen economy for a sustainable development:State-of-the-art and technological perspectives[J].J Power Source,2001, 100:171-87.
[2]Dincer I.Technical,environmental and exergetic aspects of hydrogen energy systems[J].Int J Hydrogen Energy, 2002,27:265-85.
[3]Fernandez-Moreno J,Guelbenzu G,Martín A J,et al.A portable system powered with hydrogen and one single air breathing PEM fuel cell[J].Appl Energy,2013,109:60-6.
[4]Danilova M M,Fedorova Z A,Zaikovskii V I,et al. Porous nickel-based catalysts for combined steam and carbon dioxide reforming of methane[J].Appl Catal B, 2014,147:858-63.
[5]Sun N,Wen X,Wang F,et al.Catalytic performance and characterizationofNi-CaO-ZrO2catalystsfordry reforming of methane[J].Appl Surf Sci,2011,257:9169-76.
[6]Gunn D J,El-Bousif?M A.Production of hydrogen-rich gases from steam reforming of methane in an automatic catalytic microreactor[J].Int J Hydrogen Energy,2004,29: 14278.
[7]Chalupka KA,Jozwiak WK,Rynkowski J,et al.Partial oxidation of methane on NixAlBEA and NixSiBEA zeolite catalysts:Remarkable effect of preparation procedure and Ni content[J].Appl Catal B Environ,2014,146:227-36.
[8]He H,Chen A,Lv H,Dong H,et al.Fabrication of TiO2 ?lmwithdifferentmorphologiesonNianodeand application in photoassisted water electrolysis[J].Appl Surf Sci,2013,266:126-31.
[9]Stiegel GJ,Ramezan M.Hydrogen from coal gasi?cation: An economical pathway to a sustainable energy future[J]. Int J Coal Geol,2006,65:173-90.
[10]Ashok J,Subrahmanyam M,Venugopal A.Development of methane decomposition catalysts for COxfree hydrogen[J]. Catal Surv Asia,2008,12:229-37.
[11]Awadallah A E,Aboul-Enein A A,Aboul-Gheit A K. Effect of progressive Co loading on commercial Co-Mo/ Al2O3catalyst for natural gas decomposition to COx-free hydrogen productionand carbon nanotubes[J].Energy Convers Manage,2014,77:143-151.
[12]Wang G W,Jin Y,Liu G J,et al.Production of hydrogen and nanocarbon from catalytic decomposition of methane over a Ni-Fe/Al2O3catalyst[J].Energy Fuels,2013,27: 4448-4456.
[13]Anjaneyulu C,Kumara V V,Bhargava S K,et al. CharacteristicsofLa-modifiedNi-Al2O3andNi-SiO2catalysts for COx-free hydrogen production by catalytic decomposition of methane[J].J Energy Chem,2013,22: 853-860.
[14]Zielinski J.Morphology of nickel/alumina catalysts[J].J Catal,1982,76:157-163.
[15]Scheffer B,Molhoek P,Moulijn J A.TemperatureprogrammedreductionofNiO-WO3/Al2O3Hydrodesul-phurization catalysts[J].Appl Catal,1989,46:11-30.
[16]张玉红,熊国兴,盛世善,等.NiO/γ-Al2O3催化剂中NiO与γ-Al2O3间的相互作用[J].物理化学学报,1999,15(8): 735-741.
Effect of different pre-reduction situation on performance of the catalyst for decomposition of methane
LIANG Wei,CHEN Chen,LI Teng,CHEN Xiao-bo,SHAN Hong-hong
(Sate Key Laboratory of Heavy Oil Processing,College of Chemical Engineering,China University of Petroleum,Qingdao 266580,China)
A Ni/Al2O3catalyst(w(Ni)=50%)was prepared by the sol-gel method,and the effect of reduction conditions of the catalyst on its activity on methane catalytic cracking was investigated.The fresh and spent catalysts were characterized by N2physical adsorption,XRD and H2-TPR.The results showed that,with the increase of reduction time,the conversion of methane increased firstly,and reached the highest at 90min,then reduced a little,while the selectivity of hydrogen increased gradually;with the increase of reduction temperature,the conversion of methane increased firstly,then reduced,while the selectivity of hydrogen increased gradually,reaching nearly 100%at above 800℃.Characterization results indicated that there were three kinds of NiO in the fresh catalyst,in which free NiO was the easiest to be reduced,dispersed NiO was a little harder to be reduced,and fixed NiO was the most difficult to be reduced.The activity decrease of catalyst was attributed to the sintering of reduced large-grained Ni0. Increasing reduction time was in favor of NiO reducing,however,NiO couldn’t be completely reduced by increasing time only. Increasing reduction temperature could lead to a complete reduction of NiO,but the sintering became serious.Selecting the appropriate reduction conditions would be beneficial to the catalyst to show higher activity.
methane;catalytic cracking;hydrogen;catalyst;reduction time;reduction temperature
TQ116.2;TQ426;O643.3
:A
:1001-9219(2016)01-15-06
2015-05-07;基金项目:中央高校基本科研业务费专项资金资助(No.14CX06050A),国家自然科学基金重点项目(No.U1462205);作者简介:梁威(1990-),男,硕士研究生,研究方向为石油与天然气加工,电邮liangweiupc@163. com;*
陈小博(1981-),男,博士,副教授,电邮chenxiaobo@upc.edu.cn。
