我国医疗器械分类监管改革的现状及思考
2016-02-10郭世富母瑞红李静莉
郭世富,黄 颖,母瑞红,李静莉
国家食品药品监督管理总局医疗器械标准管理中心;中国食品药品检定研究院,北京市,100050
我国医疗器械分类监管改革的现状及思考
【作者】郭世富,黄 颖,母瑞红,李静莉
国家食品药品监督管理总局医疗器械标准管理中心;中国食品药品检定研究院,北京市,100050
该文介绍了我国医疗器械分类监管的情况,对影响医疗器械的风险因素进行分析,结合我国医疗器械分类监管的现状分析,提出改革和完善我国医疗器械分类监管体制的建议。
医疗器械;分类监管;改革
经过近二十年的努力,我国初步建立了与国际接轨的医疗器械分类监管机制,实现了医疗器械产品的科学监管,保证了医疗器械产品的安全有效。然而,随着我国医疗器械产业的发展和公众对医疗健康产品的预期要求的不断提高,现在的医疗器械分类监管体系面临严峻的挑战。特别是新技术、新产品的不断涌现,新的生产方式的出现,及医疗体制改革的迫切需要,都亟待需要改革目前的分类监管体制,应对目前的各种监管难题,确保医疗器械产品的安全有效性、公众及时可获得性。
1 我国医疗器械的分类监管历程
随着市场经济的发展,我国自1989年开展医疗器械市场准入工作,以确保上市产品的安全有效。特别是2000年,国务院颁布《医疗器械监督管理条例》后,标志着我国的医疗器械监管进入了法制化的轨道。2013年,我国对《医疗器械监督管理条例》进行了修订,我国医疗器械监管进入一个新阶段[1]。由于我国幅员辽阔,而医疗器械产品跨专业广,品种繁多,我国医疗器械产品采取分级分类管理方式,以提高监管效率。按照医疗器械的风险程度,将医疗器械分为三类进行管理,不同类别的产品采取不同的管控措施。围绕医疗器械注册管理,先后发布了包括《医疗器械分类规则》、《医疗器械分类目录》等部门规章[2]。2012年发布细化6823、6830、6831、6834等4 个子目录。2013 年,我国制定发布《体外诊断试剂分类子目录》。2014 年发布《第一类医疗器械产品目录》。2015年发布了修订的《医疗器械分类规则》,细化并对有源、无源、侵入、植入、作用时间等影响医疗器械风险的因素进行了表述和定义,并以附表的形式对各种情形下产品的管理类别进行了限定[3]。
我国的医疗器械监管体系借鉴了欧盟的分类规则制和美国的目录制度,实施分类规则指导下的分类目录制度,同时发布一系列分类界定规范性文件作为补充,进一步明确了几千个产品的管理类别,实现了覆盖整个产品品种的分类监管体系。
2 影响医疗器械分类等级的产品风险因素分析
任何医疗产品都有风险,医疗器械产品在给人们带来收益的同时,也存在潜在风险。对医疗器械实现科学合理分类管理的关键在于基于风险管理的理念,实现对器械的风险辨识,根据器械风险程度,给以合理的分类,采取合理的风险控制措施,保证收益大于风险。
医疗器械的分类等级基于其对患者或使用者引起的潜在伤害,要充分考虑其预期用途和使用的技术(作用部位、作用时间、是否向患者输出能量等)。影响医疗器械风险的因素很多,美国FDA风险管理专家认为医疗产品的风险类型和风险源如图1所示。

图1 医疗器械风险类型和风险源Fig.1 Risk type and resource of medical device
大多数与医疗产品使用相关的伤害和死亡由已知的副作用(known side effects)产生。有些副作用是不可避免的,但是其他的是可以通过对产品的谨慎选择和使用来降低和防止。可避免的不良事件来源于医疗或设备差错(medication or device errors)。比如在标签、产品使用说明书中存在错误或缺陷,导致使用者不恰当的使用,或由于使用者对器械的不熟悉,对试剂错误存放等。还有可能是医疗器械性能、功能故障或损坏。即使产品在按要求的条件下使用时,仍然发生故障或损坏,不能按照预期达到所期望的功能。有一类风险源来自于产品缺陷(product defects),受现有的科学技术条件、认知水平、生产工艺、使用材料等因素的影响,其可能会产生一定程度的产品缺陷。风险可以通过在产品生产期间对产品质量控制和质量保证得以减少。
还有一些风险是不可预见的风险(remaining uncertainties)。医疗器械上市前研究存在局限性。医疗器械在上市前都必须做一系列的安全性评价,包括物理评价、化学评价、生物学评价和临床评价。目前的生物学试验都依赖于动物模型,而材料在动物体内出现的组织反应,在人体内不一定出现同样的反应。临床评价过程由于受时间、临床评价例数、个体差异,受试范围、认知水平等的影响,许多潜在的风险可能没有发现。
比如在对产品上市前审批时,对产品的临床试验,样本有限,时间短,罕见的副作用和长期结果(既包括正面的和负面的)可能不被得知。当产品投入市场以后的时候,这些人群在使用时,副作用可能被发现。甚至是一种产品在长期使用以后,不可预见的风险仍然存在。
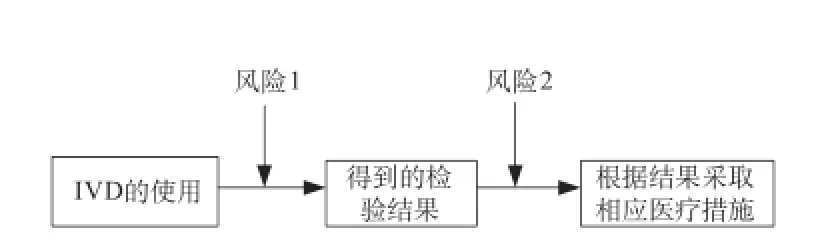
图2 体外诊断医疗器械风险分析Fig.2 IVD risk analysis
例如针对IVD的风险因素分析,如图2所示。IVD在使用时有可能失误(此为风险1),此错误的结果可能伤害到受检者(此为风险2)。只有当风险1和风险2都为零时,病患才是安全的。因此IVD的分级制度应同时考虑风险1和风险2。从IVD出错的概率来看,操作或判断愈复杂或困难的IVD,其风险就愈高。例如,IVD的科技含量愈高,技术复杂,操作者受到的挑战就愈高,因此就愈容易出错。有时虽然技术复杂度不高,但操作可能容易出错,存在做出错误的报告的风险。因此,体外诊断产品应该从错误发生率及后果的严重度来考虑,得到理想的IVD风险分级。
3 我国医疗器械分类监管现状分析
由于医疗器械产品的复杂性,我国借鉴国际通用做法,从管理角度将医疗器械分为三类管理:第Ⅰ类是指通过常规管理足以保证其安全性、有效性的医疗器械。如听诊器、外科用手术器械(刀、剪、钳)、刮痧板等。第Ⅱ类是指对其安全性和有效性应当加以控制的医疗器械。如心电图机、血压计、体温计、避孕套等。第Ⅲ类是植入人体,用于支持、维持生命或对人体具有潜在危险,对其安全性、有效性需要严格控制的医疗器械。如心脏瓣膜、心脏起搏器、支架、骨板、疝气植入物、缝合线等。我国采用集权和分权相结合的监管方式,高风险的Ⅲ类由国家总局部门的集中审评审批,对于中风险的Ⅱ类由省局审评审批,对于低风险的Ⅰ类由原先的审批变为备案管理。
对于医疗器械的界定,新修订的《医疗器械监督管理条例》对医疗器械的定义进行了重新明确。这一定义明确了我国医疗器械监管的产品范围,与国际上通常的法规定义是一致的。从这个定义可以看出,界定医疗器械要把握三个特征:一是生产商预期将这些物品直接或间接作用于人体;二是其对人体体表或体内的预期作用不是通过药理学、免疫学或代谢的手段获得,但是可能有这些手段参与并起了一定的辅助作用;三是具有明确的一个或多个特定医疗目的。同时,判断某个产品是否医疗器械,还要考虑其作用机理是否有科学的理论支持,预期用途是否能够得到足够的科学验证。如颈椎枕,宣称能治疗颈椎病,但可能其对疾病的预防、治疗、缓解作用机理不明确、无法验证等原因,在我国仍不作为医疗器械管理。
我国借鉴国外监管方式,以分类规则结合分类目录方式,较好地实现了医疗器械分类监管。但是,由于受法律地位、层级结构、处理方式等因素的影响和制约,及医疗器械产业快速发展,种类日益繁多,产生了许多问题,例如相同的产品在不同地方出现不同类别的审批结果,分类目录不能覆盖新出现的产品,分类界定申请数量剧增,国家局对《分类目录》补充的文件不断增加,诸如此类问题,经常令企业和监管人员感到困惑。
对我国2012年至2015年的审批产品数据库进行研究统计,我国审批的医疗器械产品在各子目录的分布情况如表1所示,因数据较多,只取数量排名前十的分类列表。由表1中可以看出,低风险的产品集中于“6806口腔科手术器械”、“6810矫形外科(骨科)手术器械”、“6840临床检验分析仪器”及“6864医用卫生材料及敷料”等。中度风险的产品在各个子目录的分布比较分散,其中“6822医用光学器具、仪器及内窥镜设备”、“6826物理治疗及康复设备”、“6840临床检验分析仪器”和“6863口腔科材料”、“6864医用卫生材料及敷料”和“6866医用高分子材料及制品”子目录产品比较多。而高风险产品主要分布在“6846植入材料和人工器官”、“6877介入器材”、 “6822医用光学器具、仪器及内窥镜设备”子目录等。
对各个子目录的分布进行分析发现,已上市的产品分布比较分散,其中“6831 医用X射线附属设备及部件”、“6832医用高能射线设备”、“6833医用核素设备”分布产品数量比较少。“6870软件产品”数量少,主要集中于Ⅱ类产品。
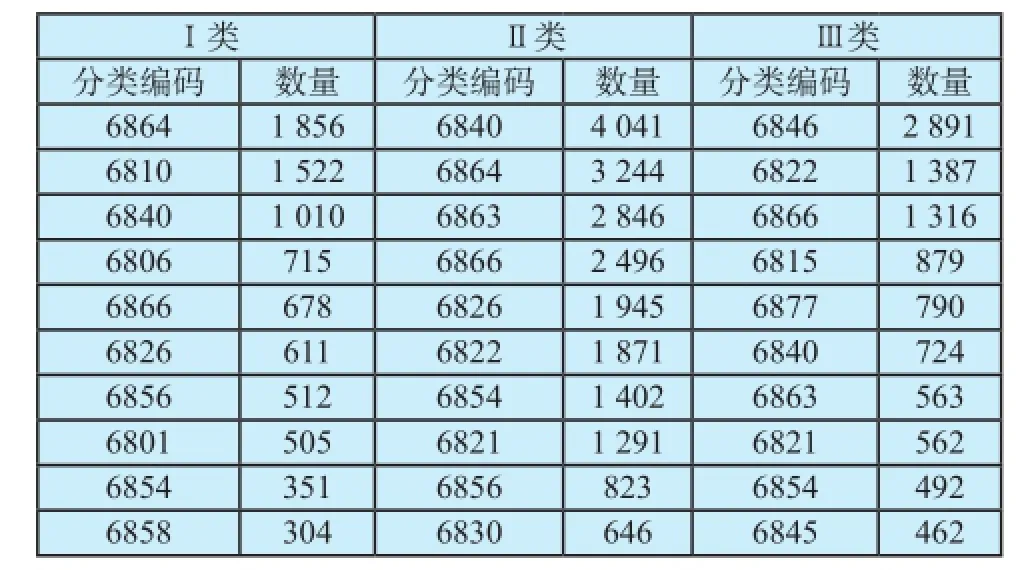
表1 上市产品在分类目录中分布情况Tab.1 Product classif cation distribution
为研究上市产品中风险程度的分布情况,对Ⅰ类、Ⅱ类、Ⅲ类产品数量的占比进行统计,如图3所示。
由图3中可以看出,我国医疗器械产品管理类别主要集中在Ⅱ类(占54%),Ⅲ类产品占比25% ,要高于Ⅰ类产品的占比21% 。Ⅱ、Ⅲ类产品总和占比高达79%。如此多的产品都要进行上市前的审评审批,按照目前的审评审批机制,大部分要经过检验检测、体系核查和临床试验验证等程序,必将造成行政资源的浪费,特别是目前在食品药品监管体制改革的大背景下,监管数量大与监管资源缺乏的矛盾会更加突出。

图3 各类别器械占比分布图Fig.3 Classif cation ration of Chinese medical device
4 关于我国医疗器械分类监管体制改革的意见和建议
当前我国正在深化医药卫生体制改革,医疗器械分类监管改革是医药体制改革重要组成部分。医疗器械的分类是医疗器械监管工作的基础,直接关系监管效率及器械产业的发展及公众对器械的可获得性。笔者根据多年技术审批工作实践和参与分类目录修订实践,谈一下完善我国医疗器械分类监管体系的意见和建议。
4.1 建立科学合理的分类目录框架
要建立基于医疗器械风险管理理念的的分类监管体系,完善细化《分类目录》。美国FDA分类目录制中,将约一千七百多种不同类型的器械按照其风险划分为三类,并归纳为16个模块[5]。各类别的数量及分布情况如表2所示。
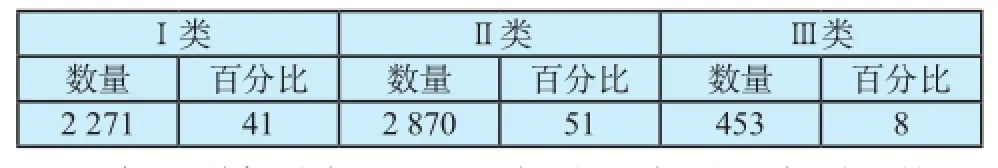
表2 FDA分类分布及占比情况 [n(%)]Tab.2 FDA classif cation distribution and ration of medical device [n(%)]
在21联邦法规862-892部分可发现,类别是按照不同医疗专业科室进行划分的。例如:829部分为放射医疗产品,882部分为神经系统产品等。而我国的医疗器械分类目录43个子目录中一部分是按照科室进行分类,如6809泌尿肛肠外科手术器械,6804眼科手术器械。而另一部分按工程特性进行分类,如6866医用高分子材料及制品,6864医用卫生材料及敷料等。这样容易造成产品交叉和遗漏,不能很好地覆盖所有的医疗器械产品。同时随着产业和技术发展,出现了许多新的产品形式,如药械产品、组合产品和辅助生殖产品等。仅包含简单分类和产品举例的《分类目录》已远不能满足我国医疗器械的分类监管需求。
因此需要汇总整理所有上市的医疗器械产品,对产品的结构组成、技术特性、预期用途进行分析、归纳、总结,借鉴欧美的分类管理经验,逐步形成符合我国监管实际的分类目录框架,保证品名举例的准确、统一、协调,使产品在目录中的定位更加准确,避免理解上的差异,保证框架的科学合理,使其更具实用性和操作性。
4.2 根据我国多年医疗器械监管实践,科学合理实施部分产品管理类别的调整
当前,我国医疗器械产品中,II、III类产品所占比重较大。根据我国医疗器械产品管理类别的分布情况,我国的医疗器械产品主要集中在II类(占54%),高于FDA(51%);III类所占的比例25%比FDA(8%)要高。所以在监管实践中,要根据根据产品的安全性和有效性,对于风险较低且可控,研发技术比较成熟的产品,放宽要求,适当降低其管理类别,科学的消减II、III类产品的数量,有效的节约行政资源,促进医疗产品的可获得性。
4.3 加强分类目录内容的研究,做到规范统一协调
目前,由于我国缺乏统一的医疗器械产品的标准术语,因此有些上市的医疗器械产品存在“不准确、不规范、不系统”的问题。例如对体表实施电刺激后收集记录电信号的仪器,可能被叫做“诱发电位系统”、“肌电诱发电位仪”、“肌电生物反馈仪”等,引起了大量的同名异物,不利于信息数据的交换[6]。再如有些夸大功效,如**病治疗仪等。有些对名称的细化程度和描述角度不同,造成同样的产品具有不同的名字。这些都为医疗器械分类目录的修订造成很大困难,也为产品的上市前审评和上市后监管造成隐患和障碍。
因此,需要对目前的分类文件进行归纳总结。同时,对近几年已上市的6万多个注册产品信息进行同名排序,合并梳理。可以借鉴FDA产品分类数据库、GMDN数据库、JMDN数据库等国际上相对成熟的产品标准术语库,结合我国医疗器械国家行业标准,详细描述产品的预期用途、组成材质,给出典型的品名举例,形成详尽的目录形式。同时,建立信息系统平台,对目录产品关联相关的所需监管措施,技术审评指南,国行标、注册产品信息等内容。这样不但有利于企业和监管机构准确判断产品分类检索,而且可以便于对上市产品进行比较,对医疗器械产品可以实施实质性等同审查,保证审查尺度,节约行政资源,对上市后产品形成监督。有利于我国公平、公正、透明的医疗器械监管体系的形成。
医疗器械分类管理医疗器械监管法规体系的基础。立足于我国的监管实际,借鉴国外的经验和成功做法,逐步建立并不断完善符合国际惯例,具有可行性、易操作性和国际协调型分类管理法规体系,是我国食品药品监管部门面临的紧迫任务。
[1] 中华人民共和国国务院. 中华人民共和国国务院令 第650号 医疗器械监督管理条例[R]. 2014-02-12.
[2] 国家药品监督管理局. 国药监械[2002]302号 医疗器械分类目录[R]. 2002-08-28.
[3] 国家食品药品监督管理总局. 国家食品药品监督管理总局令 第15号 医疗器械分类规则[R]. 2015-07-14.
[4] FDA. Managing the risks from medical product use[R].2009-05
[5] FDA. CFR - Code of Federal Regulations Title 21[EB/OL].2015-8-21. http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/ cfrsearch.cfm?cfrpartfrom=862&cfrpartto=892.
[6] 岳伟. 对医疗器械命名规则和分类目录的见解[J]. 中国医疗器械杂志, 2010, 34(1): 44-46.
Present Situation and Thinking on the Reform of the Classif cation and Regulation of Medical Devices in China
【Writers】GUO Shifu, HUANG Ying, MU Ruihong, LI Jingli Center for Medical Device Standardization Administration, CFDA; National Institute for Food and Drug Control, Beijing, 100050
This paper introduces the domestic device regulatory classif cation. Risk factors of medical equipment are analyzed. Combined with the analysis of the present situation of medical device regulatory classif cation in china, this paper puts forward advice to reform and improve China's medical device regulatory classif cation system.
medical device, classif cation, reform
F203
A
10.3969/j.issn.1671-7104.2016.05.011
1671-7104(2016)05-0355-04
2016-07-04
郭世富,E-mail:1909677524@qq.com
