阿尔茨海默氏病新易感基因的研究
2015-12-27黄旭程王莹黄智慧
黄旭程,王莹,黄智慧
(温州医科大学,浙江 温州,325035,1.检验医学院;2.基础医学院)
·综 述·
阿尔茨海默氏病新易感基因的研究
黄旭程1,王莹1,黄智慧2
(温州医科大学,浙江 温州,325035,1.检验医学院;2.基础医学院)
在阿尔茨海默氏病(AD)新易感基因领域研究中,经典的基于连锁基因和候选基因的关联研究已经逐渐被外显子测序、全基因组测序(针对孟德尔型AD)和全基因组关联研究(针对非孟德尔型AD)等替代。通过新技术寻找到的新易感基因有助于研究潜在的疾病机制。除了大样本检测新风险因素外,新一代测序方法可以对很小数量的患者进行检测。今后的研究重点将更注重转化医学的研究、单个患者的测序和个体患者生物材料的收集,这些将成为遗传子研究的核心单位。当然这一转变需要遗传科学家和临床神经科医师的紧密合作。本文对AD遗传学的最新发现和应用进行综述。
阿尔茨海默氏病;新易感基因;外显子测序;全基因组测序;全基因组关联研究
预计2030年全球将有6 600万老年痴呆症患者,2050年患者人数将会高达1.15亿。阿尔茨海默氏病(Alzheimer’s disease,AD)是老年痴呆症患者的主要疾病之一,且研究表明AD患者的病情越重,其认知功能和生活自理能力越差[1]。临床诊断上可将其分为早发型(<65岁)和迟发型(>65岁)两类。AD的病理学特征是存在淀粉样Aβ肽缠结斑块以及神经细胞中高度磷酸化的微管相关蛋白tau(microtubule-associated protein tau,MAPT)。目前已知,无论是早发型还是迟发型AD都有遗传因素。迄今在早发型AD中已发现三个突变基因,这三个基因的突变奠定了淀粉样蛋白在AD中的核心作用,淀粉样蛋白通路因此成为了AD机制中研究最广泛的通路。然而,除了常染色体显性遗传的家系外,AD的遗传模式并不是简单的常规遗传方式,而是由多种遗传和环境因素综合造成的,也就是说更适合定义为一种复杂遗传背景的疾病。另一方面关于迟发型AD,研究预测其具有高达80%的遗传力[2]。而先前许多年来的研究只证明一个遗传风险因子:ApoE ε 4等位基因,明确地参与了AD的发生发展。随着科学技术的进步,大规模的全基因组关联研究项目开始出现,能同时识别整个基因组数以百万计的遗传变异,因此能迅速识别出迟发型AD中十多个风险基因[3-4]。同时这些风险基因的发现会促使人们去关注其他相关通路,如脂质代谢、免疫系统和突触功能的机制等。
通过AD全基因组关联研究的Meta分析有助于阐明更多的危险因素[3-4]。这些新一代的测序方法可以应用于某些小型家系原因不明的AD和一些不相关的患者,同时它扩大了探针的数量,有助于检出潜在的变异风险。临床应用这些先进的技术方法对患者生物材料的采样和遗传诊断筛查具有重大的意义。我们对AD遗传学研究进行了综述,着重讨论最新的发现及其应用,以及面临的挑战。
1 最新发现的遗传因子
1.1 复杂基因型AD的新风险位点 长期以来AD的遗传学研究难以进行,而自2009年以来国际间大型合作的开展已经改变了这个现象,使得近几年来AD的遗传学研究取得较大进展。通过欧洲和国际性全基因组关联的合作,已至少找到九个新的易感基因位点。加利福尼亚路德大学(California Lutheran University,CLU)发布了此结果。此外全基因组关联和复制研究已经发现补体受体1(complement receptor,CR1)、PICALM和Myc基因盒依赖的相互作用蛋白1(Myc box-dependent-interacting protein 1,BIN1)及其附近位点的单核苷酸多态性。这些基因都是在第一波的大型合作性的全基因组关联研究中检测到的,不过仍需要继续开展合作,确定在跨膜四域蛋白A(Membrane-spanning 4-domains subfamily A,MS4A)集群、CD2相关蛋白(CD2-associated protein,CD2AP)、CD33、EphA1和三磷酸腺苷结合盒转运子(ATP-bingding cassette A7,ABCA7)中的单核苷酸多态性之间的关联,AD常见的变异及其作用,见表1[3-4]。
AD国际基因组学研究小组将会继续补充此列表的内容,此项目汇集了来自全球四个最大的全基因组关联组织的数据,并用一系列方法进行了分析。例如研究影响发病年龄的遗传因素、探索基因与整个AD生理途径的相互作用等。通过与帕金森病和额颞叶变性全基因组关联研究结果的对比,发现致病因子都是全基因组关联的最常见的风险基因,但淀粉样前体蛋白(amyloid precursor protein,APP)、早老蛋白1(presenilin 1,PSEN1)和早老蛋白2(presenilin 1,PSEN2)这三个常见变异并不是复杂型AD的危险因素[4-5]。
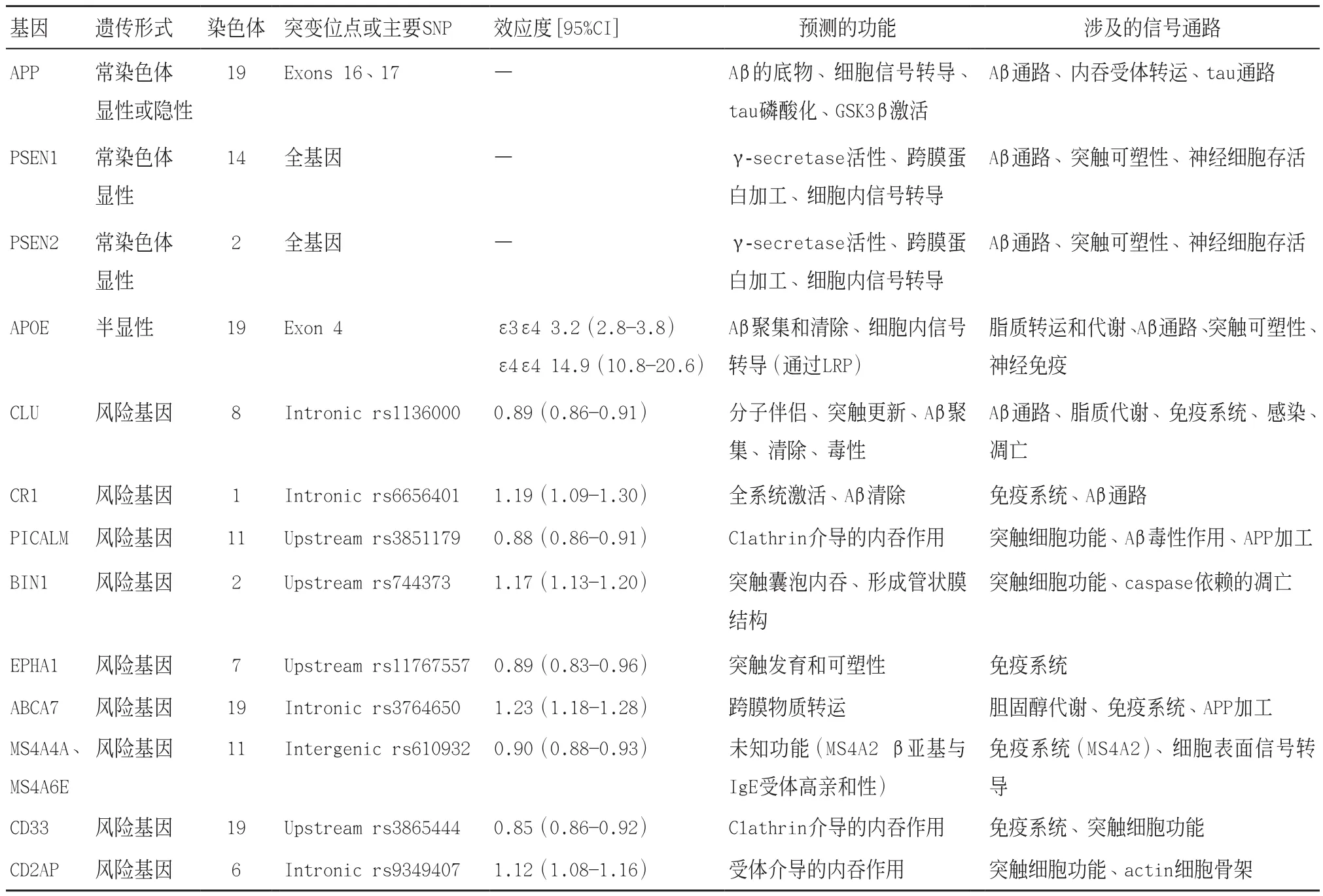
表1 孟德尔形式和非孟德尔形式阿尔茨海默氏症间的因果关系和风险基因
1.2 早发型AD和孟德尔型AD的风险基因重检 新一代DNA测序技术的进步给人类基因组测序带来了前所未有的改革,这使研究者们开始重新审视早发型或家族性AD患者的遗传子研究,尤其是对于传统的连锁分析研究来说规模太小的家庭。对新一代DNA测序技术来说,仅仅对几个相关患者全外显子或全基因组的测序比较就足以揭示孟德尔疾病新的致病突变。因此对AD的分子遗传子来说,其核心应是单个患者而不是家系。到目前为止,已报道了两个AD患者的全外显子测序研究。第一个研究意外发现NOTCH3(Homo sapiens notch 3)上的错义突变与AD有关,这个基因以前一直被认为与常染色体显性脑动脉病伴皮层下梗死和白质脑病有关[6],因此并没有作为家系的先证者被筛查出来。同样,通过对肌萎缩侧索硬化症患者进行全外显子测序,并在临床、病理和遗传学方面与额颞叶变性相互结合研究,揭示了某意大利家庭常染色体显性遗传的VCP突变[7]。VCP突变以前曾在包涵体肌病、Paget氏病和额颞叶痴呆症患者中被检测到[7]。
我们知道单个基因的突变可以解释一系列神经退行性疾病的表型,如AD的MAPT Arg406Trp突变、皮克病的PSEN1 Gly183Val突变、AD和帕金森病的颗粒体蛋白(Granulin,GRN)的无义突变和错义突变、AD的C9orf72重复扩增等,这一发现至今仍具有重要的临床意义,为遗传原因不明的AD患者进行所有已知神经退行性基因的诊断筛查提供了根据。由于神经退行性疾病相关基因的数量太大,通过Sanger测序来进行诊断显然不能实现,而新一代测序技术能够同时筛选所有参与神经退行性疾病的基因。因此,这种新技术将大大有助于神经退行性疾病的个性化治疗,特别是使针对目标分子引起的治疗方式变得可行。
第二个在早发型AD患者中进行全外显子测序的研究表明:在分拣蛋白相关受体-1(sortilinrelated receptor receptor 1,SORL1)上存在错义突变和无义突变[8],它编码一种神经元性整合蛋白能结合APP,并将其定位至内涵体回收通路。尽管缺乏SORL1变异体功能性的证据,但编码SORL1变异体在AD中的高频出现尤其值得注意,因为这个基因先前被认为是复杂型AD的一个危险因素。这个发现意味着一个基因的致病变异与常规风险变异都可能在疾病中发挥作用。
全外显子测序是最常被用来研究单基因遗传孟德尔疾病的方法,主要是因为与全基因组测序相比,外显子组只占整个基因组1%~2%;不仅成本低,而且大多数序列变化所导致的严重表型效应都只位于基因组的编码部分。然而越来越多的证据表明,非编码区的变异,如肌萎缩性侧索硬化症的内含子可溶性超氧化物歧化酶(superoxide dismutase1,SOD1)突变,AD的启动子变异也会增加神经退行性疾病的风险。此外,全基因组测序比全外显子测序提供了更好的外显子覆盖范围。随着测序成本的下降和生物信息学技术的推进,全基因组测序将来会广泛应用。有研究通过对1 795个冰岛人全基因组测序数据的研究,在β-分泌酶(β-site APP cleaving enzyme 1,BACE1)酶切位点附近发现一个保护性的错义突变(ala676thr),与那些没有这个突变的人相比,AD患者淀粉样肽形成有所降低,并有更好的认知能力[9]。这一发现支持了一种观点,即降低BACE1的剪切能够预防疾病,并可能减少淀粉样蛋白形成和减弱淀粉样蛋白毒性作用。
全基因组测序研究仍面临着巨大挑战,因为与一个外显子组(有20 000~50 000单核苷酸多态性)相比,一个基因组约有300万,因此进行验证和跟踪的基因变异数量相当巨大。由于这需要大范围收集临床和生物化学表型并进行随访,与临床医生密切合作显得尤为重要。目前开展的大型合作,如显性遗传性阿尔茨海默网络(DIAN)联盟[10]旨在了解罕见的单基因形式的AD,还有欧洲早发型老年痴呆症协会[11]针对早发型AD患者开展临床与遗传转化研究,这两个组织的研究工作将在今后发挥重要作用。
2 从间接相关到遗传病因的确定
2.1 潜在的疾病变异的研究 全基因组关联研究已经证实了常见的遗传变异对于复杂AD的作用。风险变异的存在为疾病的病理机制提供了新思路。然而,在这些风险基因位点的关联变异往往没有出现明显的功能性疾病和连锁不平衡现象(两个或多个基因位点的等位基因的非随机联系)。最相关的单核苷酸多态性位于成簇蛋白(Clusterin,CLU)上,例如某一个内含子的突变起初被认为并不影响CLU的表达或功能,但事实上是CLU最相关的单核苷酸多态性[4]。
仔细研究全基因组关联位点对于鉴定潜在的风险等位基因很有必要。不仅仅是检测最显著的全基因组相关的单核苷酸多态性,而且也应研究在这些位点完整的遗传变异,包括常见的、罕见的变异和拷贝数变异。至今,大多数遗传因子的后续研究都是基于第一波全基因组关联的基因,即CLU、CR1、PICALM和BIN1,只有少数基于第二波全基因组关联基因。
特别是CLU,很多研究已经证实了其与AD的相关性,是全基因组相关单核苷酸多态性中最显著的一个[12]。一个常见的多态性连锁不平衡(显著单核苷酸多态性)已被认为是可能的功能变异。然而其他的研究并没有重复出现此相关性,表明这种多态性不能或至少不完全能解释在CLU上的全基因组关联性。有研究正在重新对CLU编码的区域进行不同程度的测序,以确认可能的致病变异。最大的一项研究发现,在AD患者中,CLU的β-链外显子上存在罕见致病变种聚集模式,其次是等位基因频率<5%、存在非同义替换、9个碱基的插入或缺失,暗示此蛋白亚单位在疾病风险中发挥作用[13]。
与CLU相似,CR1的首次研究也局限于全基因组相关单核苷酸多态性的结果。此单核苷酸多态性位于非编码序列,没有明显的功能相关性。CR1位点的特点是序列高度重复,这使对遗传危险因素的真实性验证产生了一定困难。但通过对AD具有易感性的CR1拷贝数的个体变异的检测找到了AD和功能性拷贝数变异之间的关联。此功能性的拷贝数决定CR1蛋白的长度,并决定了在补体级联C3b或C4b和辅因子结合位点的数目[14]。与此拷贝数变异联合可以解释早期全基因组关联研究中所发现的单核苷酸多态性。
而对于四种新发现的AD基因位点,与其关联的最强信号都在基因上游或下游,这使确定致病变异的难度有所增加,见表1。以PICALM为例,全基因组关联信号是基因的远上游,这一发现在后续研究中得以验证[15],但不一定是编码PICALM的变异。同样,在散发性、家族性迟发型AD[15]和全基因组关联研究的Meta分析[3]中,证实了与BIN1的上游具有关联性。但强的关联并不一定意味着这些单核苷酸多态性的因果关系,一个基因的上游非编码DNA变异也可能对疾病表型有直接的作用,例如在胆固醇的代谢中,分拣蛋白(sortilin 1,sort1)sort1启动子上游120 kb的一个非编码变异就能调控该基因的表达。
早先的Sanger测序需耗费大量劳力,且不适用于在基因组非编码区识别遗传风险因素。相比之下,新一代测序技术能检测出参与疾病或对疾病有影响的常见的、罕见的变异以及拷贝数变异,这些检测已开始应用在复杂性疾病的研究中,难度是要找到合适的算法和足够大的样本数据来进行分析。这种结合新一代测序技术、算法和足够的样本数据的分析方法,已运用于鉴定与AD相关联的罕见CLU变异,并证实了这一策略具有卓越的应用价值[13]。此外,新一代测序方法也证实了NCSTN上的罕见变异和迟发型AD之间具有一定的关联[16],此变异也与家族性早发型AD相关[17]。一项功能性遗传学研究调查了不同nicastrin单倍体,提出了不同单倍体和NCSTN的单核苷酸多态性在常见AD中的功能差异[18]。一些罕见的变异可能会积累并最终超过疾病的易感性阈值,由于AD的风险基因如CLU[13]可能是罕见的致病变异,这种在等位基因范围内的转变需要对所有新的风险基因作进一步的详细或者针对性的再测序。对于与复杂AD无关的患者,新一代测序研究将在致病基因和风险变异的识别上发挥至关重要的作用。功能学研究将进一步鉴定这些变异的致病作用。
2.2 遗传变异的生物学效应 那么这些检测出的遗传变异是否都具有生物学效应,且其生物学效应都集中在哪些方面呢。事实上,AD的九个新风险基因在功能上具有一定的相关性,大多数都与脂质传递假说(CLU和ABCA7)、补体系统、炎症免疫系统(CLU、CR1、ABCA7、CD33和EphA1)和细胞突触功能(如细胞内吞作用PICALM、BIN1、CD33和CD2AP)相关[19],见表1。MS4A集群可能作用于信号传导,具体功能未知。虽然还不能确定其发病机制,但可以据此作出猜想。例如clusterin是一种多功能蛋白质,其疾病相关的变异可能会影响一系列的功能,包括脑内淀粉样Aβ42的清除、细胞凋亡、胆固醇转运和炎症反应[20]。同样,CR1变异能激活补体反应并减少大脑中淀粉样斑块的形成。PICALM基因的遗传变异可能影响突触功能,因为PICALM基因能够调节突触小泡相关膜蛋白(vesicle-associated membrane protein,VAMP2)转运,这对突触小泡的融合及学习记忆是至关重要的[21]。因此AD易感基因并不是随机发现的,而是基于对AD全基因组关联数据的信号通路分析得出的,即用生物信息学方法对参与疾病易感性的信号通路进行分析,同时结合全基因组测序中单核苷酸多态性的重要性来共同发现新的信号途径[19]。目前科学家通过结合生物学预测、功能学分析和患者生物标本研究,来验证AD的变异模式。研究主要集中在CLU和CR1,见图1。
第一,调查疾病相关性变异是否影响基因产物的量(在RNA转录和蛋白质翻译水平),及其是否可以作为疾病的早期标志物或潜在的治疗靶点。有意思的是发现AD基因在调控区与拷贝数变异区具有很强的关联性。几项研究已检测出CLU蛋白浓度以期判断CLU的蛋白浓度是否可以用作生物标记物[22],同时检验遗传变异是否与信使RNA和蛋白质浓度相关[22-23]。研究发现AD患者脑血浆和脑脊液中的CLU浓度会明显增加[24],但蛋白表达和基因水平的变异并不完全一致。下一步需对更大样本量的AD患者展开研究,开展完整的CLU基因序列并重点关注其异构体,也许能揭示AD风险因子的变异如何真正调控CLU的表达。
第二,深入认识疾病的发病机制,研究已检测在血液、脑脊液和脑中,遗传变异对淀粉样蛋白Aβ 1-42和tau(t-tau和p-tau181)的作用。对CLU、CR1和PICALM的研究指出,PICALM能降低脑脊液中淀粉样蛋白Aβ 1-42浓度,CR1单核苷酸多态性能增加脑脊液中β淀粉样蛋白Aβ 1-42浓度[14]。但AD脑脊液生物标志物和全基因组关 联研究中,并没有发现与生物标志物浓度相关的新基因位点[25]。因此尚没有明确的证据显示基因型与β-淀粉样蛋白或tau蛋白的表型相关,见表2。这或许是由于研究设计或小尺度效应的问题,长期以来妨碍了AD候选基因的研究,将来真正的风险等位基因的发现可能会得到更强相关性的结果。
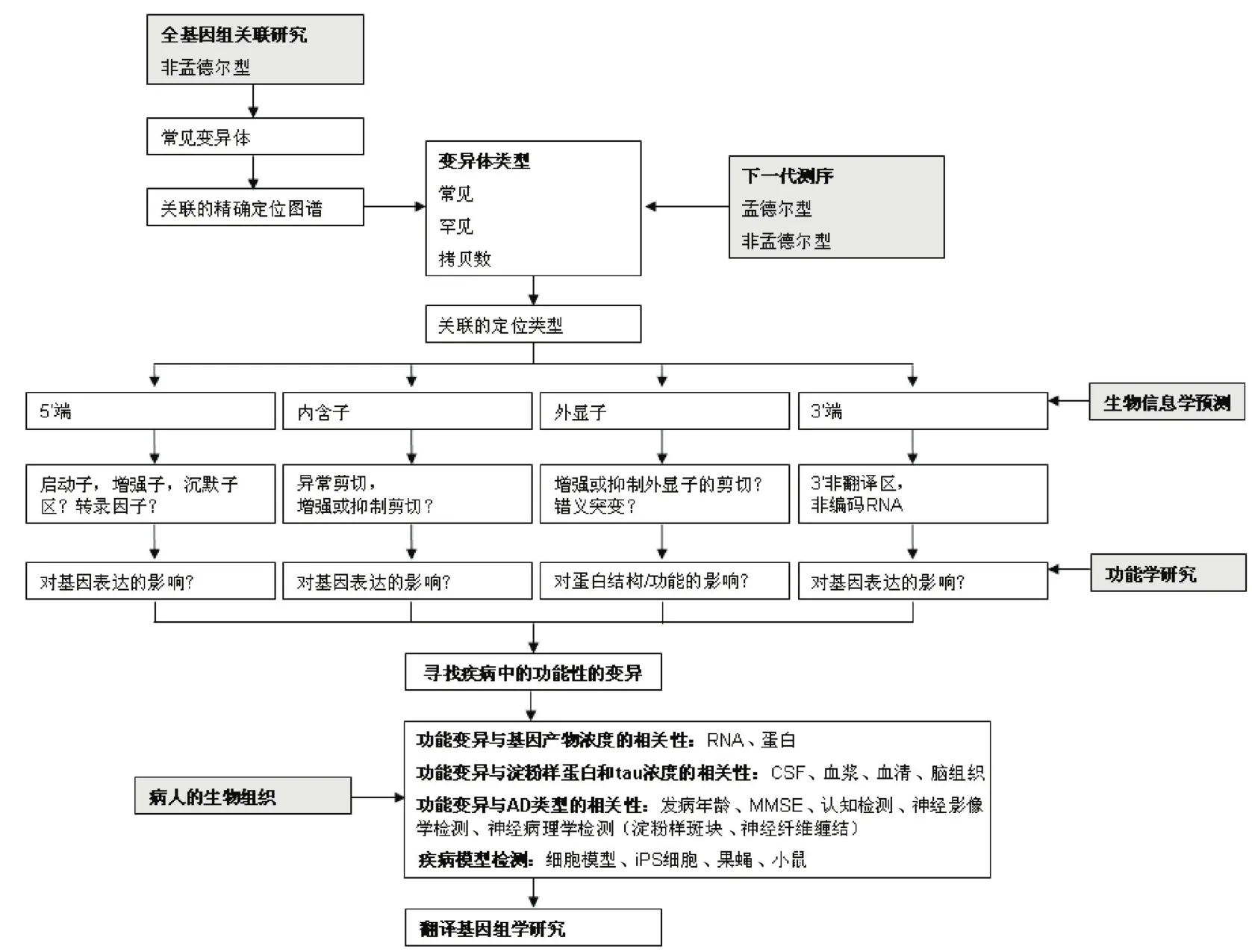
图1 对AD的遗传相关性、潜在的功能性变体和生物学功能的鉴定
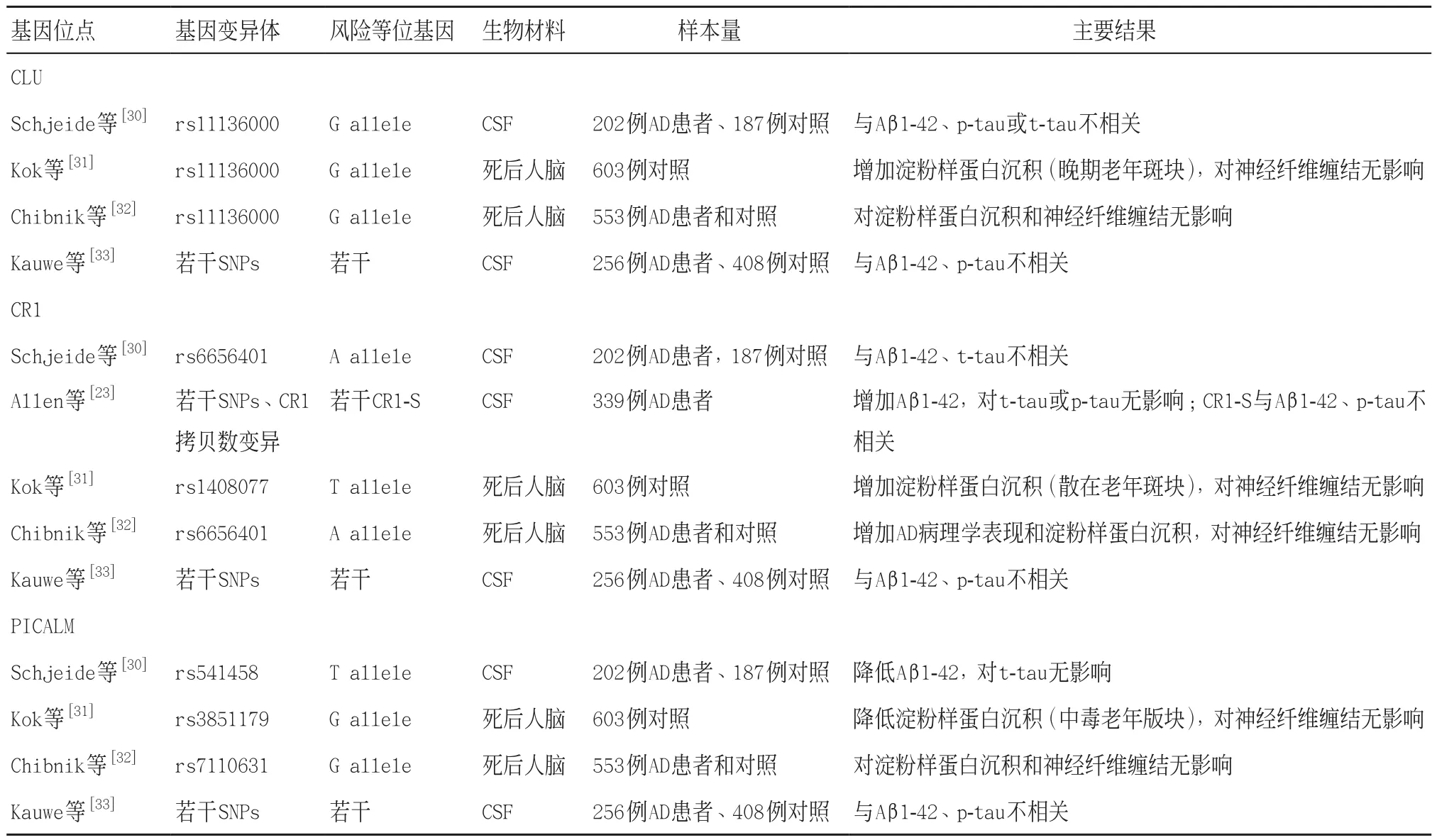
表2 第一波全基因组关联基因研究发现与阿尔茨海默病相关的生物标志物
第三,通过模式生物研究揭示未知的相关性。例如通过检测几种新基因在Aβ和tau通路中的作用,证实了在酵母、线虫和大鼠皮层神经元中,clathrin介导的内吞基因PICALM、BIN1、CD2AP与淀粉样β蛋白毒性效应有直接关系[26]。此外,通过模式生物(如果蝇)的功能筛选可以得出人类全基因组关联研究的结果。通过在体观察tau转基因果蝇眼睛的表型,来研究该基因对AD tau的神经毒性[27]。因此,今后应对所有显著或者可能相关的基因在模式生物系统上进行功能检测,观察其对tau和Aβ毒性的作用,这为可能的作用机制提供了依据。
第四,结合相关遗传表现与内在表型数据分析致病机制。分析相关遗传表现与内在表型数据(如神经影像学特征、发病年龄、精神状态和认知能力)之间的关系有助于研究疾病机制。基因位点(CLU、CR1、PICALM和BIN1)对神经影像学相关指标(如内嗅皮质厚度)和AD神经病理学的影响表明:这些性状至少部分地取决于这些位点的基因组序列,见表3。如果大样本量能重复此结果,会使这种联系得到进一步验证。
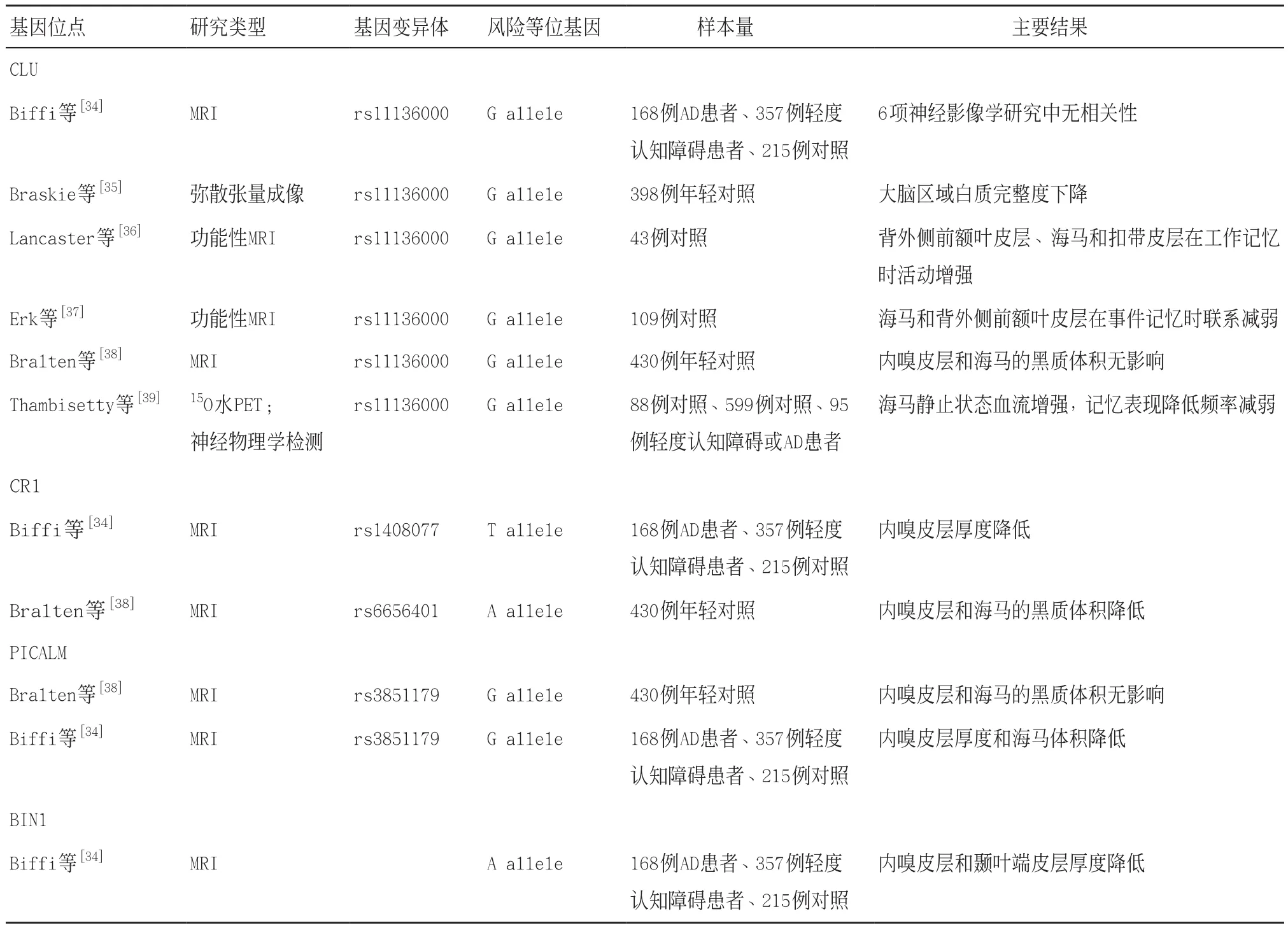
表3 若干第一波的全基因组关联基因的神经影像学研究
通过全基因组关联研究寻找遗传变异对AD神经影像学表型的影响和生物标志物的浓度等的研究已经开展,但新发现的AD全基因组关联基因几乎没有与影响影像学表型的基因重叠,除了除了发现PICALM风险等位基因rs3851179与内嗅皮质厚度的增加有关[28]。一项正在开展的AD神经影像学研究(Neuroimaging Initiative)将会收集和验证AD疾病中的生物标记物的所有数据,包括血检和脑脊液检测,同时结合MRI和PET扫描,将有助于找到遗传变异和神经影像学表型之间的联系。采用多国合作的大样本合并全基因组单核苷酸多态性和结构性MRI数据的方法(经Meta分析联合神经影像遗传学研究)有效地确定了遗传变异与海马、颅内、全脑体积之间的关联[29]。此外,国际AD基因组学研究项目(IGAP)的一系列Meta分析(临床、认知检测、影像学及生物标志物)将有助于确定影响这些性状的遗传变异。一旦在风险基因中检测到潜在遗传变异,就能更好地证明其与内表型的相关性。
3 展望
高通量基因组技术的出现已经改变了阿尔茨海默氏病的遗传变异的研究模式,大队列的全基因组关联研究使得人们对复杂的遗传形式有了深入的认识,至今已经发现9个具有小尺度效应的风险基因。随着新一代测序方法的应用,包括假说自由的方式(全基因组和全外显子组测序)和假说驱动的方式(针对全基因组关联的候选基因再测序)将有助于AD的检测和开发靶点治疗药物。
[1] 麻小莉, 董乐丹, 傅晔, 等. 中重度阿尔茨海默病患者的神经心理学症状研究[J]. 温州医学院学报, 2010, 40(6): 536-538.
[2] Gatz M, Reynolds CA, Fratiglioni L, et al. Role of genes and environments for explaining Alzheimer disease[J]. Arch Gen Psychiatry, 2006, 63(2): 168-174.
[3] Hollingworth P, Harold D, Sims R, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease[J]. Nat Genet, 2011, 43(5): 429-435.
[4] Lambert JC, Heath S, Even G, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease[J]. Nat Genet, 2009, 41(10): 1094-1099.
[5] Van Deerlin VM, Sleiman PM, Martinez-Lage M, et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions[J]. Nat Genet, 2010, 42(3): 234-239.
[6] Guerreiro RJ, Lohmann E, Kinsella E, et al. Exome sequencing reveals an unexpected genetic cause of disease: NOTCH3 mutation in a Turkish family with Alzheimer’s disease[J]. Neurobiol Aging, 2012, 33(5): 1008-1023.
[7] Johnson JO, Mandrioli J, Benatar M, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS[J]. Neuron, 2010, 68(5): 857-864.
[8] Pottier C, Hannequin D, Coutant S, et al. High frequency of potentially pathogenic SORL1 mutations in autosomal dominant early-onset Alzheimer disease[J]. Mol Psychiatry, 2012, 17(9): 875-879.
[9] Jonsson T, Atwal JK, Steinberg S, et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline[J]. Nature, 2012, 488(7409): 96-99.
[10] Bateman RJ, Xiong C, Benzinger TL, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease [J]. N Engl J Med, 2012, 367(9): 795-804.
[11] van der Zee J, Gijselinck I, Dillen L, et al. A pan-European study of the C9orf72 repeat associated with FTLD: geographic prevalence, genomic instability and intermediate repeats[J]. Hum Mutat, 2013, 34(2): 363-373.
[12] Ross OA, Soto-Ortolaza AI, Heckman MG, et al. Association of LRRK2 exonic variants with susceptibility to Parkinson’s disease: a case-control study[J]. Lancet Neurol , 2011, 10(10): 898-908.
[13] Bettens K, Brouwers N, Engelborghs S, et al. Both common variations and rare non-synonymous substitutions and small insertion/deletions in CLU are associated with increased Alzheimer risk[J]. Mol Neurodegen, 2012, 7: 3.
[14] Brouwers N, Van CC, Engelborghs S, et al. Alzheimer risk associated with a copy number variation in the complement receptor 1 increasing C3b/C4b binding sites[J]. Mol Psychiatry, 2011, 17(2): 223-233.
[15] Lambert JC, Zelenika D, Hiltunen M, et al. Evidence of the association of BIN1 and PICALM with the AD risk in contrasting European populations[J]. Neurobiol Aging, 2011, 32 (4): 756-765.
[16] Lupton MK, Proitsi P, Danillidou M, et al. Deep sequencing of the nicastrin gene in pooled DNA, the identification of genetic variants that affect risk of Alzheimer’s disease[J]. PLoS One, 2011, 6(2): e17298.
[17] Dermaut B, Theuns J, Sleegers K, et al. The gene encoding nicastrin, a major gamma-secretase component, modifies risk for familial early-onset Alzheimer disease in a Dutch population-based sample[J]. Am J Hum Genet, 2002, 70(6): 1568-1574.
[18] Hamilton G, Killick R, Lambert JC, et al. Functional and genetic analysis of haplotypic sequence variation at the nicastrin genomic locus[J]. Neurobiol Aging, 2012, 33(8): 1848.
[19] Karolien Bettens, Kristel Sleegers, et al. Genetic insights in Alzheimer’s disease[J]. Lancet Neurol, 2013, 12(1): 92-104.
[20] Nuutinen T, Suuronen T, Kauppinen A, Salminen A. Clusterin: a forgotten player in Alzheimer’s disease[J]. Brain Res Rev, 2009, 61(2): 89-104.
[21] Harel A, Wu F, Mattson MP, Morris CM, Yao PJ. Evidence for CALM in directing VAMP2 traff cking[J]. Traffc, 2008, 9(3): 417-429.
[22] Schurmann B, Wiese B, Bickel H, et al. Association of the Alzheimer’s disease clusterin risk allele with plasma clusterin concentration[J]. J Alzheimers Dis, 2011, 25(3): 421-424.
[23] Allen M, Zou F, Chai HS, et al. Novel late-onset Alzheimer disease loci variants associate with brain gene expression[J]. Neurology, 2012, 79(3): 221-228.
[24] Schrijvers EM, Koudstaal PJ, Hofman A. Breteler MM. Plasma clusterin and the risk of Alzheimer disease[J]. JAMA, 2011, 305(13): 1322-1326.
[25] Kim S, Swaminathan S, Shen L, et al. Genome-wide association study of CSF biomarkers Abeta1-42, t-tau, and ptau181p in the ADNI cohort[J]. Neurology, 2011, 76(1): 69-79.
[26] Treusch S, Hamamichi S, Goodman JL, et al. Functionallinks between Abeta toxicity, endocytic traffcking, and Alzheimer’s disease risk factors in yeast[J]. Science, 2011, 334 (6060): 1241-1245.
[27] Shulman JM, Chipendo P, Chibnik LB, et al. Functional screening of Alzheimer pathology genome-wide association signals in Drosophila[J]. Am J Hum Genet, 2011, 88(2): 232-238.
[28] Furney SJ, Simmons A, Breen G, et al. Genome-wide association with MRI atrophy measures as a quantitative trait locus for Alzheimer’s disease[J]. Mol Psychiatry, 2010, 16 (11): 1130-1138.
[29] Stein JL, Medland SE, Vasquez AA, et al. Identifcation of common variants associated with human hippocampal and intracranial volumes[J]. Nat Genet, 2012, 44(5): 552-561.
[30] Schjeide BM, Schnack C, Lambert JC, et al. The role of clusterin, complement receptor 1, and phosphatidylinositol binding clathrin assembly protein in Alzheimer disease risk and cerebrospinal fuid biomarker levels[J]. Arch Gen Psychiatry, 2011, 68(2): 207-213.
[31] Kok EH, Luoto T, Haikonen S, Goebeler S, Haapasalo H, Karhunen PJ. CLU, CR1 and PICALM genes associate with Alzheimer’s-related senile plaques[J]. Alzheimers Res Ther, 2011, 3(2): 12.
[32] Chibnik LB, Shulman JM, Leurgans SE, et al. CR1 is associated with amyloid plaque burden and age-related cognitive decline[J]. Ann Neurol, 2011, 69(3): 560-569.
[33] Kauwe JS, Cruchaga C, Karch CM, et al. Fine mapping of genetic variants in BIN1, CLU, CR1 and PICALM for association withcerebrospinal fluid biomarkers for Alzheimer’s disease[J]. PLoS One, 2011, 6(2): e15918.
[34] Biff A, Anderson CD, Desikan RS, et al. Genetic variation and neuroimaging measures in Alzheimer disease[J]. Arch Neurol, 2010, 67(6): 677-685.
[35] Braskie MN, Jahanshad N, Stein JL, et al. Common Alzheimer’s disease risk variant within the CLU gene aff ects white matter microstructure in young adults[J]. J Neurosci, 2011, 31(18): 6764-6770.
[36] Lancaster TM, Baird A, Wolf C, et al. Neural hyperactivation in carriers of the Alzheimer’s risk variant on the clusterin gene[J]. Eur Neuropsychopharmacol, 2011, 21(12): 880-884.
[37] Erk S, Meyer-Lindenberg A, Opitz von Boberfeld C, et al. Hippocampal function in healthy carriers of the CLU Alzheimer’s disease risk variant[J]. J Neurosci, 2011, 31(49): 18180-18184.
[38] Bralten J, Franke B, Arias-Vasquez A, et al. CR1 genotype is associated with entorhinal cortex volume in young healthy adults[J]. Neurobiol Aging, 2011, 32(11): 2106.e7-11.
[39] Thambisetty M, Beason-Held LL, An Y, et al. Alzheimer risk variant CLU and brain function during aging[J]. Biol Psychiatry, 2013, 73(5): 399-405.
(本文编辑:吴彬)
R394
CDOI: 10.3969/j.issn.2095-9400.2015.03.018
2014-04-21
温州医科大学2013年度校级学生科研立项课题资助(wyx201301035)。
黄旭程(1993-),男,浙江金华人,本科生。
黄智慧,副教授,Email:hzhzju021@163.com。
