胆道闭锁肝纤维化研究进展
2015-12-21丁美云詹江华
丁美云,詹江华
胆道闭锁肝纤维化研究进展
丁美云1,詹江华2△
胆道闭锁(BA)是严重危及婴幼儿生命健康的消化系统疾病之一,晚期肝脏纤维化是导致患儿死亡的主要原因。在胆道闭锁发病过程中,病毒感染可诱导一系列免疫和炎症反应,导致调节性T细胞(Treg细胞)减少,CD14表达增高,多种炎症通路以及转化生长因子-β(TGF-β)/Smad2/3促纤维化通路被激活。激活的通路产生大量炎性介质损伤肝细胞和胆管细胞,释放各种促炎因子、氧代谢产物和细胞因子,进一步加重肝、胆系统损伤造成肝细胞内环境失衡,失衡的内环境伴随肝实质细胞、肝巨噬细胞、肝内聚集的炎性细胞等发生适应性变性、坏死、增生,导致肝星形细胞(HSCs)激活,HSCs转化为成纤维细胞,促进肝纤维化进程。免疫、炎症损伤、促纤维化通路是导致胆道闭锁肝纤维化肝硬化的三大重要因素。
胆道闭锁;肝硬化;T淋巴细胞,调节性;转化生长因子β;抗原,CD14;Smad蛋白质类
胆道闭锁(biliary atresia,BA)目前仍然是小儿外科中最严重的肝胆系统疾病之一,具体发病原因尚不十分清晰。目前认为,在胚胎发育期机体内先天发育异常可以诱发BA;而另外一种病因是在围生期,患儿肝胆系统受到病毒等多种因素损伤后发生一系列固有和适应性免疫反应以及炎症反应,导致促炎因子、CD14等大量炎性因子聚集;同时发生调节性T细胞(Treg细胞)增殖失衡以及转化生长因子(TGF)-β促纤维化通路激活,它们共同作用逐渐破坏肝胆系统、肝实质,形成促进BA肝纤维化发展的特殊内环境,导致肝实质及肝内外胆管损伤进行性加重并促进肝硬化形成[1]。不可逆转的肝纤维化、肝硬化是BA疾病进展最严重的结局,是影响BA预后的重要因素,本文拟对BA肝脏纤维化研究综述如下。
1 免疫炎性损伤与促进肝纤维化
目前大多认为BA是由于机体感染病毒引发固有或适应性免疫反应,导致炎症介质增多、Treg细胞减少,同时CD14高表达,使胆管结构受到破坏或发育停滞,引起免疫和炎症反应损伤胆管上皮细胞、肝细胞等,并促进肝细胞释放各种促炎因子、细胞因子。各种促炎性因子、免疫产物促进BA肝纤维化发展[2]。
1.1 炎症介质激活肝星形细胞(HSCs)诱导肝纤维化病毒感染激活由核因子(NF)-κB介导的炎症反应引起胆管上皮脱落,脱落的胆管上皮被肝巨噬细胞吞噬提呈至T细胞受体,激活T细胞使之分化成细胞毒性T细胞,细胞毒性T细胞通过分泌炎性介质如促炎因子白介素(IL)-2、干扰素(IFN)、细胞因子[3]以及激活Fas/FasL系统两个方面攻击BA胆管上皮细胞。进而机体发生炎症反应,炎性细胞如淋巴细胞、自然杀伤细胞等浸润进一步造成胆管损伤、胆汁淤积,同时肝内鹅去氧胆酸(CDC)增多损伤肝细胞导致肝细胞坏死、调亡。损伤的肝细胞和各种细胞因子可以刺激HSCs活化,激活的HSCs增生并迁移至门管区产生大量基质成分,增生的HSCs同时经历表型转化形成肌成纤维细胞[4],肌成纤维细胞分化促进胆道纤维化、肝纤维化、肝硬化形成[2]。
与此同时,病毒抗原通过生成IFN-γ激活特异性T细胞,引发胆道上皮组织炎性反应并刺激巨噬细胞释放一氧化氮、肿瘤坏死因子(TNF)等多种细胞因子,这些细胞因子、病毒和坏死产物共同作用继续加重胆管上皮细胞、肝细胞坏死、凋亡,致使肝细胞内环境严重失衡,这种失衡状态反过来进一步活化HSCs,加重肌成纤维细胞以及基质成分的形成,促进肝脏胆管纤维化发生[5]。
各种炎症通路被病毒激活后产生多种炎性介质,炎性介质损伤肝脏和胆管引发炎症反应,炎症反应又继续加重肝脏和胆管损伤促进炎性介质生成,它们作用于HSCs形成肌纤维细胞等,促进肝组织纤维化。因此,炎性损伤在肝纤维化进程中产生巨大作用,炎性介质以及各种细胞因子、坏死物质形成对肝纤维化发展起重要推进作用。
1.2 Treg细胞减少促进肝纤维化发展Treg细胞对于维持免疫内环境稳定性非常重要,它与效应性辅助性T细胞新亚群Th17细胞密切相关,都是从最初的CD4+T细胞前体分化而来。Treg细胞在感染性疾病和自身免疫性疾病中扮演着重要角色,其通过产生调节性细胞因子,接触抑制及竞争必需细胞因子产生作用,具有广泛的免疫抑制作用。Th17细胞与炎症反应性疾病,自身免疫性疾病有关。它主要通过分泌IL-17α发挥功能,调节组织炎性因子浸润以及激活NF-κB介导的炎症反应,同时IL-17α诱导促炎细胞因子如IL-6、TNF、单核细胞趋化蛋白(MCP)-1、巨噬细胞炎性蛋白(MIP)-2等的表达,导致肝组织和胆管组织损伤。然而促进Th17细胞增殖的炎性通路干扰Treg细胞发挥免疫抑制功能,即Treg细胞阻止炎性反应的功能会被Th17细胞增殖破坏。在BA实验和临床研究中都有文献证实Treg细胞数量缺失,其数量缺失使免疫抑制作用降低或消失进而抑制炎症反应能力减弱,从而导致促炎性反应迅速放大,以至形成无法控制的炎性反应[6]。研究发现,BA患儿外周血Th17细胞显著增加而Treg细胞数量明显减少,伴随BA肝纤维化加重,Treg/Th17细胞比率进一步降低[6]。Treg和Th17细胞之间平衡的紊乱见于原发性胆汁性肝硬化、自身免疫性硬化性胆管炎,这说明在BA中Treg/Th17细胞比率降低可能是免疫炎症性紊乱导致肝纤维化发展潜在的病理机制。
对BA肝脏细胞因子的研究发现IL-1β、IL-6、IL-32和TGF-β1在BA患儿外周血表达增加,这些因子能够促进Th17细胞和其他效应细胞增多,提示在BA患儿肝实质内存在一个促炎症微环境[7]。BA动物模型已经证明TGF-β1影响Treg和Th17细胞的发展,低水平TGF-β1、IL-6和IL-21联合作用促进Th17细胞分化,而分化的Th17细胞分泌大量IL-17α[8]。据此推测BA患儿体内可能存在TGF-β1介导Th17细胞分化,它们共同作用加重免疫炎症损伤从而促进肝纤维化,同时TGF-β自身也是介导肝纤维化发展的重要因子。
2 胆道闭锁肝组织TGF-β促纤维化通路及作用
2.1 TGF-β在胆道闭锁肝纤维化形成中的作用TGF-β在BA患儿大部分由肝内胆管上皮细胞产生,与BA的肝脏纤维化关系密切[9]。TGF-β促纤维化在BA肝纤维化进程中起重要作用,主要通过Smad家族蛋白调节,Smad蛋白家族是TGF-βs超家族重要的细胞因子,由8种不同的蛋白Smad1~8构成,通过可逆磷酸化对多种信号传导通路的功能起着关键的调节作用[10]。
在促纤维化过程中,TGF-β通过整合素蛋白αvβ6等因素启动,基于与其受体的联系诱导Smad2/3磷酸化,磷酸化后形成与Smad4的络合物。这个络合物向细胞核移动,启动包括细胞外基质(ECM)蛋白的基因转录,增加ECM蛋白的表达,同时也抑制了抗纤维化蛋白酶抑制剂[11]。TGF-β下游基质金属蛋白酶1组织抑制因子(TIMP)-1和人纤溶酶原激活物抑制因子(PAI)-1过度表达加速肝脏纤维化进程[12],见图1。目前研究证实其作用机制主要通过以下两方面来完成:一是通过抑制各种基质金属蛋白酶减少ECM降解,同时刺激HSC/ECM络合物,激活HSCs等,促进肝纤维化进程;另一方面通过抑制抗纤维化蛋白酶抑制剂促进组织纤维化。研究证实,在啮齿动物中阻断Smad2/3信号通路可以阻止肝脏纤维化,提示阻止这一信号通路是治疗胆道闭锁肝纤维化的可能方法[9,13]。研究发现Smad相关蛋白(SIP)-1是一个通过绑定激活Smads 1、2、3、5、8与TGF-β路径相互作用的转录因子[14];Smad-3和SIP-1相伴随增加,SIP-1绑定激活的Smads家族和Smad络合物引起靶基因转录激活物减少[15]。因此SIP-1伴随TGF-β路径而产生并且与其作用相反。TGF-β/Smad2/3通路在BA肝纤维化中起重要作用,与ECM以及纤维细胞的合成、增殖作用有关,但TGF-β通路对BA肝胆炎症有何作用有待进一步研究。
2.2 TGF-β1加速肝纤维化并导致肝功能衰竭TGF-β有6种不同的亚型TGF-β1~6,其中TGF-β1是肝纤维化发生过程中起关键作用的细胞因子。TGF-β1来源于Kuffer细胞,显著刺激HSC/ECM络合物,它作为潜在纤维化细胞因子以广泛调节作用包括HSC激活、ECM蛋白化合物诱导和基质金属蛋白酶抑制作用为大家熟知[16]。HSCs被TGF-β1激活后在肝损伤部位增生并迁移至门管区产生大量基质成分,HSCs又可以分泌TGF-β1、上调TGF-β1受体,形成自分泌循环。TGF-β1能够刺激纤维细胞合成与增殖,以及一些细胞外基质成分诸如胶原蛋白Ⅰ、Ⅲ、Ⅳ和纤维黏连蛋白(fibronectin)分泌。TGF-β1在小叶中心部的表达明显高于肝门区域[14],它在BA晚期过度表达,在启动复杂的纤维化进程中起到重要作用,并加速肝脏纤维化进程。有研究指出存在TGF-β1/Smad2/3促纤维化路径,且TGF-β1通路是引起BA肝脏纤维化的重要通路之一[13]。
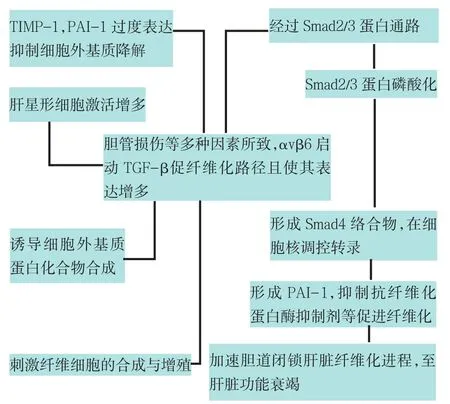
Fig.1 The pathway of fibrosis induced by TGF-β图1 TGF-β促纤维化路径
3 CD14加速BA肝纤维化发展
3.1 CD14结构CD14主要结构特征为N-末端富含亮氨酸的重复单位,人类CD14氨基酸序列中39~44位区段是结合脂多糖(LPS)的必须区段,该结构可能在识别LPS中发挥关键作用,CD14分子以膜CD14(mCD14)和可溶CD14(sCD14)2种形式存在[17]。mCD14是一个50~55 ku的受体,通过糖基磷酸化肌醇(glycosyl-phosphatidyl inositol,GPI)固定于细胞表面,主要在巨噬细胞、树突状细胞、单核细胞和中性粒细胞的表面表达,sCD14是一种糖蛋白,由单核细胞产生,但血浆中sCD14的主要来源是肝实质细胞。
3.2 CD14在BA的双重性在BA肝内炎细胞表面检测到mCD14,说明mCD14在炎细胞表面表达并且参与炎性反应,而它在肝内Kuffer细胞和肝窦内皮细胞的表达显示:CD14高表达促进炎症反应,促进NF-κB表达,造成机体免疫反应激活和全身炎症反应综合征,导致胆汁淤积、肝脏损伤[18]。LPS对肝脏具有损伤作用,在BA肝损害中作用尤为明显。通过LPS方式诱导的Kuffer细胞、中性粒细胞、肝实质细胞和胆道上皮细胞中都有CD14表达,而Kuffer细胞可以修饰已经内吞的LPS,然后将其传递到肝实质细胞继而分泌到胆汁中[18]。这说明CD14在吸收和清除LPS的循环途径中有一定的作用。高水平CD14通过炎症反应损伤肝脏甚至增加LPS诱导的死亡率,同时CD14也具有协同Kuffer细胞共同转运LPS从而减轻肝脏损害的作用,因此BA中CD14在不同水平具有双重性。
肝细胞直接分泌sCD14,这可以增加对内毒素的清除,在BA早期胆汁淤积时血浆中sCD14含量增加,提示sCD14在肝脏能够阻止由于胆汁淤积导致的内毒素增加,减轻肝脏损伤[17,19]。相反,BA晚期sCD14减少却没有伴随肝脏和血中内毒素减少,据此推测Kuffer细胞内缺少保障CD14传递LPS作用持续进行的机制,而导致炎症反应传递,造成不可逆转的肝损伤和晚期BA患者肝组织纤维增生、肝硬化的结局[20]。
3.3 CD14促肝纤维化通路CD14在BA肝细胞内激活信号传导机制可能有蛋白酪氨酸激酶(protein tyrosine kinase, PTK)和磷脂酰肌醇(phosphatidyl inositol,PI)2个途径。PI途径通过产生第二信使来实现信号转导并且发挥细胞内生物学效应。PTK途径是CD14受体细胞内信号传递中的另一重要通路。当LPS直接激活肝细胞,损伤胆管上皮细胞时,在LPS刺激的数分钟内,一些特定的靶蛋白被酪氨酸磷酸化,从而激活促分裂原活化蛋白激酶途径,催化转录因子发生磷酸化修饰,调控相关基因的表达,导致CD14在肝细胞内表达上调。同时细菌毒素和LPS激活大量肝巨噬细胞,肝内产生大量活化CD14阳性巨噬细胞,CD14阳性巨噬细胞产生大量细胞因子如TGF、血小板源性生长因子(PDGF)、TNF等,这些细胞因子继续激活HSCs生成大量基质以及促肝纤维化因子,导致肝纤维化形成[21]。
内毒素等损伤因素导致CD14在BA肝脏中高表达,引起胆汁淤积、肝脏损伤,加大促炎症反应,CD14本身也可能作为炎性因子刺激机体免疫反应活化HSCs,并且促进TGF-β1分泌及其受体上调[21]。因此,CD14作为一种炎性介质不仅介导免疫反应和炎性损伤,而且CD14损伤胆管细胞、促进大量细胞因子释放的作用也可激活TGF-β促纤维化通路。因此,CD14在BA肝脏纤维化发展中起着重要的介导作用。
综上,围生期病毒等众多因素损伤机体引发炎症、免疫反应生成CD14、TGF-β等多种细胞因子,同时激活TGF-β促纤维化通路加重免疫和炎症反应,促进BA肝纤维化进程。免疫、炎症、促肝纤维化通路相互作用,是BA肝纤维化发展三大重要因素。除此之外BA肝纤维化发展是否还有其他路径,以及TGF-β促纤维化通路如何调节免疫和炎症反应,如果打断以上通路是否可以减轻BA肝胆纤维化程度仍需进一步研究。
[1]Zhan JH,Guan ZW,Zhang H.Attention to neonatal cholestasis:Enhance the early diagnostic rate of Biliary atresia[J].Chinese Journal of Applied Clinical Pediatrics,2014,29(11):803-806.[詹江华,管志伟,张辉.重视新生儿胆汁淤积:提高胆道闭锁的早诊率[J].中华实用儿科临床杂志,2014,29(11):803-806].doi:10.3760/cma.j. issn.2095-428X.2014.11.002.
[2]Antoniades CG,Khamri W,Abeles RD,et al.Secretory leukocyte protease inhibitor:a pivotal mediator of anti-inflammatory responses in acetaminophen-induced acute liver failure[J].Hepatology,2014,59 (4):1564-1576.doi:10.1002/hep.26933.
[3]Moore SW,Zabiegaj-Zwick C,Nel E.Problems related to CMV infection and biliary atresia[J].S Afr Med J,2012,102(11):890-892.doi: 10.7196/samj.6163.
[4]Kisseleva T,Cong M,Paik Y,et al.Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis[J].Proc Natl Acad Sci USA,2012,109(24):9448-9453.doi:10.1073/pnas.1201840109.
[5]Vejchapipat P,Poomsawat S,Chongsrisawat V,et al.Elevated serum IL-18 and interferon-gamma in medium-term survivors of biliary atresia[J].Eur J Pediatr Surg,2012,22(1):29-33.doi:10.1055/s-0032-1306260.
[6]Brindley SM,Lanham AM,Karrer FM,et al.Cytomegalovirus-specific T-cell reactivity in biliary atresia at the time of diagnosis is associated with deficits in regulatory T cells[J].Hepatology,2012,55(4): 1130-1138.doi:10.1002/hep.24807.
[7]Okamura A,Harada K,Nio M,et al.Interleukin-32 production associated with biliary innate immunity and proinflammatory cytokines contributes to the pathogenesis of cholangitis in biliary atresia[J]. Clin Exp Immunol,2013,173(2):268-275.doi:10.1111/cei.12103.
[8]Zhou L,Lopes JE,Chong MM,et al.TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing ROR gammat function [J].Nature,2008,453(7192):236-240.doi:10.1038/nature06878.
[9]Massague J.TGF signalling in context.Nat Rev Mol Cell Biol,2012, 13(10):616-630.doi:10.1038/nrm3434.
[10]Cheng X,Alborzinia H,Merz KH,et al.Indirubin derivatives modulate TGFβ/BMP signaling at different levels and trigger ubiquitinmediated depletion of nonactivated R-Smads[J].Chem.Biol,2012,19 (11):1423-1436.doi:10.1016/j.chembiol.2012.09.008.
[11]Markovics JA,Araya J,Cambier S,et al.Interleukin-1beta induces increased transcriptional activation of the transforming growth factorbeta-activating integrin subunit beta8 through altering chromatin architecture[J].JBiolChem,2011,286(42):36864-36874.doi: 10.1074/jbc.M111.276790.
[12]Burch ML,Zheng W,Little PJ.Smad linker region phosphorylation in the regulation of extracellular matrix synthesis[J].Cell Mol Life Sci,2011,68(1):97-107.doi:10.1007/s00018-010-0514-4.
[13]Lee JH,Lee H,Joung YK,et al.The use of low molecular weight heparin-pluronic nanogels to impede liver fibrosis by inhibition the TGF-β/Smad signaling pathway[J].Biomaterials,2011,32(5):1438-1445.doi:10.1016/j.biomaterials.2010.10.023.
[14]Iordanskaia T,Hubal MJ,Koeck E,et al.Dysregulation of upstream and downstream transforming growth factor-β transcripts in livers of children with biliary atresia and fibrogenic gene signatures[J].J PediatrSurg,2013,48(10):2047-2053.doi:10.1016/j.jpedsurg.2013.03.047.
[15]Conidi A.Few Smad proteins and many Smad-interacting proteins yield multiple functions and action modes in TGF-β/BMP signaling in vivo[J].Cytokine Growth Factor Rev,2011,22(5-6):287-300.doi: 10.1016/j.cytogfr.2011.11.006.
[16]Kamato D,Burch ML,Piva TJ,et al.Transforming growth factor-β signalling:role and consequences of Smad linker region phosphorylation[J].Cell Signal,2013,25(10):2017-2024.doi:10.1016/j.cellsig.2013.06.001.
[17]Kim D,Kim JY.Anti-CD14 antibody reduces LPS responsiveness via TLR4 internalization in human monocytes[J].Mol Immunol,2014, 57(2):210-215.doi:10.1016/j.molimm.2013.09.009.
[18]Chou MH,Chuang JH,Eng HL,et al.Endotoxin and CD14 in the progression of biliary atresia[J].J Transl Med,2010,8:138.doi:10.1186/ 1479-5876-8-138.
[19]Berenger BM,Hamill J,Stack D,et al.Membrane CD14,but not soluble CD14,is used by exoenzyme S from P.aeruginosa to signal proinflammatory cytokine production[J].J Leukoc Biol,2011,90(1):189-198.doi:10.1189/jlb.0510265.
[20]Litzman J,Nechvatalova J,Xu J,et al.Chronic immune activation in common variable immunodeficiency(CVID)is associated with elevated serum levels of soluble CD14 and CD25 but not endotoxaemia [J].Clin Exp Immunol,2012,170(3):321-332.doi:10.1111/j.1365-2249.2012.04655.x.
[21]Liu HH,Hu Y,Zheng M,et al.CD14 SNPs regulate the innate immune response[J].Mol Immunol,2012,51(2):112-117.doi:10.1016/ j.molimm.2012.02.112.
(2014-08-01收稿2014-11-14修回)
(本文编辑李国琪)
Advances in the research of liver fibrosis in biliary atresia
DING Meiyun1,ZHAN Jianghua2△
1The Graduate School of Tianjin Medical University,Tianjin 300070,China;2 Tianjin Children’s Hospital△
Biliary atresia(BA)is one of the most serious digestive system diseases,which threatens the health of infants. Liver fibrosis is a major cause of death in children with BA.In the process of the pathogenesis of BA,virus infection can induce a series of immune and inflammatory reaction,result in a decrease of regulatory T cells(Treg cells)and high expression of CD14,activating a variety of inflammatory pathways and TGF-β/Smad2/3 pro-fibrogenic pathway,which produces a large number of medium damage of liver cells and bile duct cells,releases proinflammatory factor,oxygen metabolism matter and cytokines.These changes further aggravate damage of hepatobiliary system and cause the internal environment imbalance of liver parenchyma cells.The imbalance of internal environment with adaptive degeneration and necrosis in liver parenchyma cells,hepatic macrophages and gathered inflammatory cells leads to the activation of hepatic stellate cells(HSCs).HSCs can be converted into fibroblast cells,and promote the process of liver fibrosis.Immune and inflammatory lesions,pro-fibrogenic pathway are the important factors in contributing to liver fibrosis and cirrhosis of biliary atresia.
biliary atresia;liver cirrhosis;T-lymphocytes,regulatory;transforming growth factor beta;antigens,CD14; Smad proteins
R726.574
A
10.3969/j.issn.0253-9896.2015.01.002
天津市卫计委重点攻关项目(14KG129)
1天津医科大学研究生院(邮编300070);2天津市儿童医院外科
丁美云(1988),女,硕士在读,主要从事小儿普通外科、胆道闭锁方面研究
△通讯作者及审校者zhanjianghuatj@163.com
