基于公共子序列的OPSM双聚类算法
2015-12-14傅俊橦李杰进王杜齐邝秋华张美珍
薛 云 ,傅俊橦,李杰进,王杜齐,邝秋华,张美珍,肖 化
(华南师范大学物理与电信工程学院,广州510006)
随着DNA 微阵列检测技术的发展,产生的基因表达数据量呈现爆炸性增长态势[1]. 如何对基因表达数据进行有效的分析,挖掘有用信息已经成为后基因组时代的研究热点.
作为针对基因分析的数据挖掘模型,双聚类算法应运而生[2-7].双聚类和传统聚类算法的本质区别在于,双聚类可以对基因表达数据矩阵的行和列同时进行聚类,挖掘基因表达数据中的局部信息,从而确定在1个实验条件子集下表现出相似行为的基因.这种无监督学习技术是发现基因之间共表达或共调控关系、预测基因功能、分析转录调控、阐释生物学通路和提供疾病机理等研究的重要手段[2-6].
因为多个基因在关联表达时,其各自的表达数值不需要完全相同[6],所以基因表达水平的升降模式比基因的精确表达水平更有意义. 因此,2003年Ben-Dor 等[6]提出OPSM (Order preserving submatrices)双聚类模型,其根据是基因表达水平在不同时间点或不同实验条件下表现出相同的上调或下调趋势. Fang[8-9]、Hochbaum[10]等分别提出了几种近似的OPSM 模型,诸如POPSM、AOPSM,但是这些模型不能保证OPSM 的精确度. 目前众多针对OPSM 问题的精确算法在处理巨型基因数据矩阵的问题上都存在严重缺陷:时间复杂度和空间复杂度过高.因此在执行时,在保证精确度的前提下必须限制所搜索双聚类的行列数量,这影响了部分具有重大生物意义的基因聚类(Deep OPSM 模式)的发现.本文提出一种高效的精确性算法,用于挖掘出基因数据矩阵中的OPSM,针对Deep OPSM 问题,分析并验证了算法解决该问题的优点.
1 基本概念和定义
1.1 基因表达数据集
基因表达数据可用矩阵D=(dij)m×n表示:
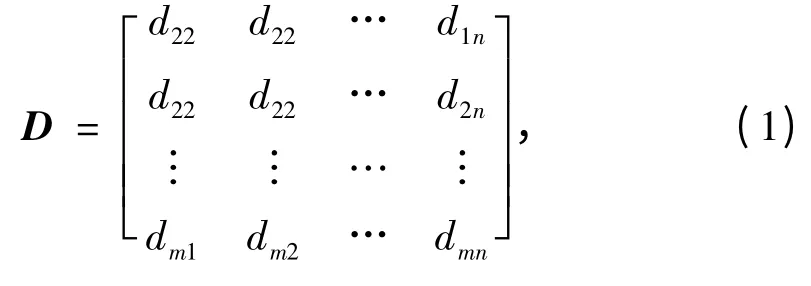
其中包含m个基因对象,n个样本,元素dij表示第i个基因在第j个条件下的表达水平值.
1.2 公共子序列
序列的子序列是指从给定字符序列中任意地(不一定连续)去掉若干个字符后所形成的序列.如果1个序列既是c1序列的子序列又是c2的子序列,则称这个序列为序列c1和c2的公共子序列.
1.3 OPSM 与Deep OPSM 的定义和意义
假设矩阵D 存在1个子矩阵,对应的行列集合为(R,C),其中R 包含于矩阵D 的行集合中,C 包含于矩阵D 的列集合中,当子矩阵(·)中每一行元素都呈现一致性变化趋势时,此时子矩阵(R,C)就称为OPSM 聚类[7].图1A 为基因g1~g6在a ~g 这7 种实验条件下的表达值(简称“七因子值”)折线图,表达值的变化趋势杂乱无章,无规律可循. 但如果将基因g1、g2、g6在a、c、d、f、g 这5 种实验条件下的表达值从矩阵中抽选值(简称“五因子值”)画出对应折线图(图1B),这时可以清晰地看出同升同降的共性,即所挖掘的OPSM[6].进一步对各行按元素值从小到大排序,则可以得出一致上升的模式(图1C).

图1 基因数据OPSM 挖掘前后的表达值图示Figure 1 Expression data of OPSM before and after mining
行数少、列数多的OPSM 称为Deep OPSM,这是OPSM 的一种特殊类型.从生物学的意义来说,Deep的意义在于即使基因数较少(2个基因),只要它们在众多的实验环境中,都具有相同变化趋势的表达值时,则判断这2个基因存在紧密联系,具有重要的生物学意义[11].过去研究大都集中在寻找大群体基因中的OPSM 聚类,但目前生物学家更感兴趣的是“小群体基因在很多实验环境下紧密地互调节关系”[12],因而更应该去关注和研究一个大基因群体里面数量很少的基因,它们极有可能在某些生物进程或疾病机理上充当着重要的角色[13],因此Deep OPSM 在基因表达数据的挖掘中理应受到更多研究.但大部分传统挖掘算法不能很好地解决Deep OPSM 问题,当挖掘大群体基因中OPSM 聚类时,如果设定较低的行阈值,将大量消耗计算机内存,甚至导致内存溢出.而如果为了减少计算机消耗而设定较高的行阈值,将导致大量的Deep OPSM 被忽略.本文提出一种新的精确算法,用于挖掘基因表达数据集,以解决这一问题,首先提出一种高效方法寻找2个序列的所有公共子序列,并结合数据结构得到全部OPSM,也包含全部Deep OPSM.
2 针对OPSM 的精确算法
首先针对基因数据矩阵中的每2 行寻找其公共子序列,然后在整个基因数据矩阵的范围内,对找到的公共子序列进行支持度统计. 当公共子序列的支持度超过设定阈值时,保存该子序列并输出为目标OPSM.
在解决时间复杂度高的问题时,利用每2个序列构造出来的匹配矩阵,快速地找到2个序列的所有公共子序列.在解决空间复杂度高的问题时,利用了C++编程环境中的STL map,存储产生的序列并统计相应的支持度.算法流程如图2 所示.
2.1 数据预处理
原始的基因表达数据矩阵中存在着部分缺失的元素,所以在数据预处理过程中必须对这部分缺失的元素进行填充.数据预处理具体操作包括:(1)将数据矩阵中缺失的元素填充为0;(2)根据基因谱中基因表达水平的大小,对各个实验条件按照表达水平递增进行排列;(3)用列标识置换数据矩阵中对应的元素.
通过以上的数据预处理操作,基因数据矩阵可以转换成1个序列数据集(表1)
此时,OPSM 的挖掘问题转化为序列模式挖掘问题,对基因数据矩阵的局部聚类即转化为寻找频繁公共子序列的问题.
2.2 寻找全部公共子序列ACS 的算法
本文提出一种高效的方法寻找2个序列的全部公共子序列即ACS (all common subsequences).
定义1 假设有2个序列c1={A B C D E F }和c2={F A B C D E },将2个序列的其中一个序列作为行标识,另一个序列作为列标识.当且仅当元素dij对应的行标识与列标识相同时,将元素dij置为相应的行(列)标识,其他元素置“0”,即组成1个矩阵M,称之为匹配矩阵(图3).
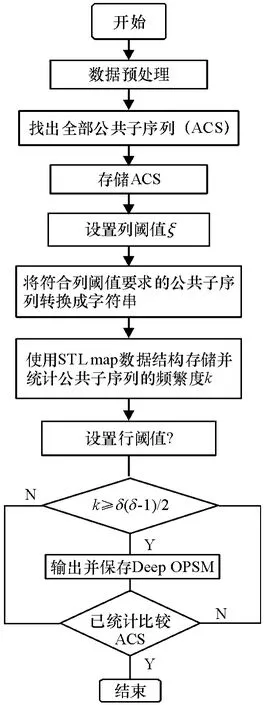
图2 算法流程图Figure 2 Flowchart of the algorithm
推论1 由序列c1以及序列c2生成的匹配矩阵M 中,当且仅当元素B 的位置在元素A 的位置的右下方(即B 行列号都大于A)时,元素B 可接在元素A 的后面构成序列c1和c2的公共子序列.
2.3 查找和存储ACS
根据匹配矩阵的定义和叙述,从匹配矩阵中寻找公共子序列的方法如下:
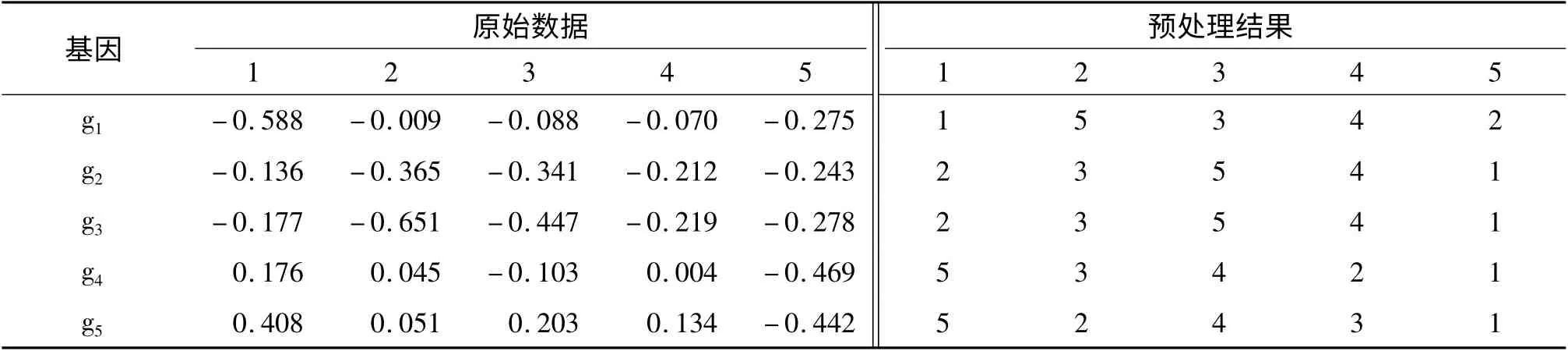
表1 数据预处理结果Table 1 Results of data preprocessing
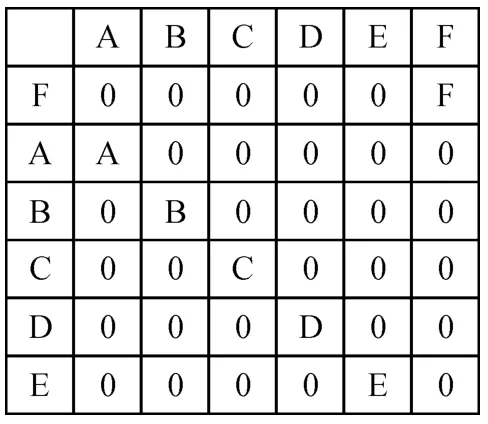
图3 匹配矩阵Figure 3 Matching matrix
扫描匹配矩阵中第一列,查找非零元素如A,此元素A 为第一个公共子序列的起点.
扫描下一列的元素,查找非零元素如B,判断该元素B 的行号:
如果元素B 的行号大于上一个元素A 的行号,则元素B 可以接在原有公共子序列的后面,形成新的公共子序列.
如果元素B 的行号小于上一个元素A 的行号,则元素B 不可以接在原有公共子序列的后面,应该生成新的公共子序列的起点.
以此类推,直到判断完匹配矩阵的最后一列,并以树的形式存储查找结果. 由匹配矩阵生成的结果存储如图4 所示.
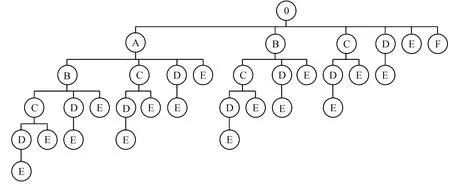
图4 存储树结构Figure 4 Structure of storage tree
遍历该树,得到2个序列的所有公共子序列.在找出每2个序列的所有公共子序列后,对结果进行存储.对于未出现的新子序列,创建新的存储单元;对于此前已经出现过的子序列,要在其行支持度记录上加1.因为子序列的个数多,所以必须使用一种适合快速检索的数据结构进行存储.
2.4 频繁项集统计方法
利用C++标准程序库中STL map 对产生的公共子序列进行存储并统计其支持度. 对比支持度与设定的最小支持度,将频繁项集作为结果输出.STL map 的底层数据结构是红黑树. 红黑树是一种自平衡二叉查找树,可以在O(lg n)时间内做查找、插入和删除,这里的n 是树中元素的数目.运用STL map对产生的公共子序列进行存储并统计支持度,能够降低算法的空间和时间复杂度.
采用STL map 对生成的子序列进行存储具有以下优点:(1)用红黑树的数据结构能快速检索元素,可以在O(lg n)时间内做查找和插入,而双聚类问题本身是NP 问题,因此该数据结构非常适用于双聚类的精确算法;(2)可以根据生成的子序列的长度进行判断,从而对子序列进行条件存储.当程序只对较长的生成子序列进行存储时,能够快速地找到长的频繁项集,这对于挖掘出Deep OPSM 非常有利;(3)根据条件存储,可以同时在多台计算机分析同1个数据集,实现并行计算,提高分析处理速度,适应大型数据矩阵的分析需求.
3 案例分析
为了验证本文方法的有效性,利用真实的基因数据矩阵来进行实验. 使用半乳糖酵母基因数据集中的时间序列,其中包含205个半乳糖酵母基因在20个时间点(大于2个完整的细胞周期)的表达数据[14].它是一组在真实的实验条件下获得的基因表达数据,是Yee 等[15]从生物数据库中搜索出的205条基因,它们分别归属于4个功能类别(表2),这4个功能类别也被广泛用于衡量聚类质量的外部标准.
系统开发平台参数如下:
处理器:Intel(R)Core(TM)i3 CPU M 380 @2.53 GHz;内存:4G;运行速度2 533.3 MHz;计算机系统为Windows7;运行软件为VS2010;实验编程语言为C++.

表2 数据集的4个功能类别Table 2 Four functions of the genes in this dataset
3.1 OPSMs 可视化分析
使用本文提出的算法挖掘出全部OPSMs,并把4个典型OPSMs 进行可视化操作,绘制成折线图(图5),直观地发现其共同特点. 横坐标为对应OPSMs 聚类的列号,纵坐标为对应的聚类元素在基因表达数据矩阵上的表达值. 每一条折线所对应的聚类基因,其表达值在原矩阵中呈现同增同减的一致变化趋势,即OPSM.
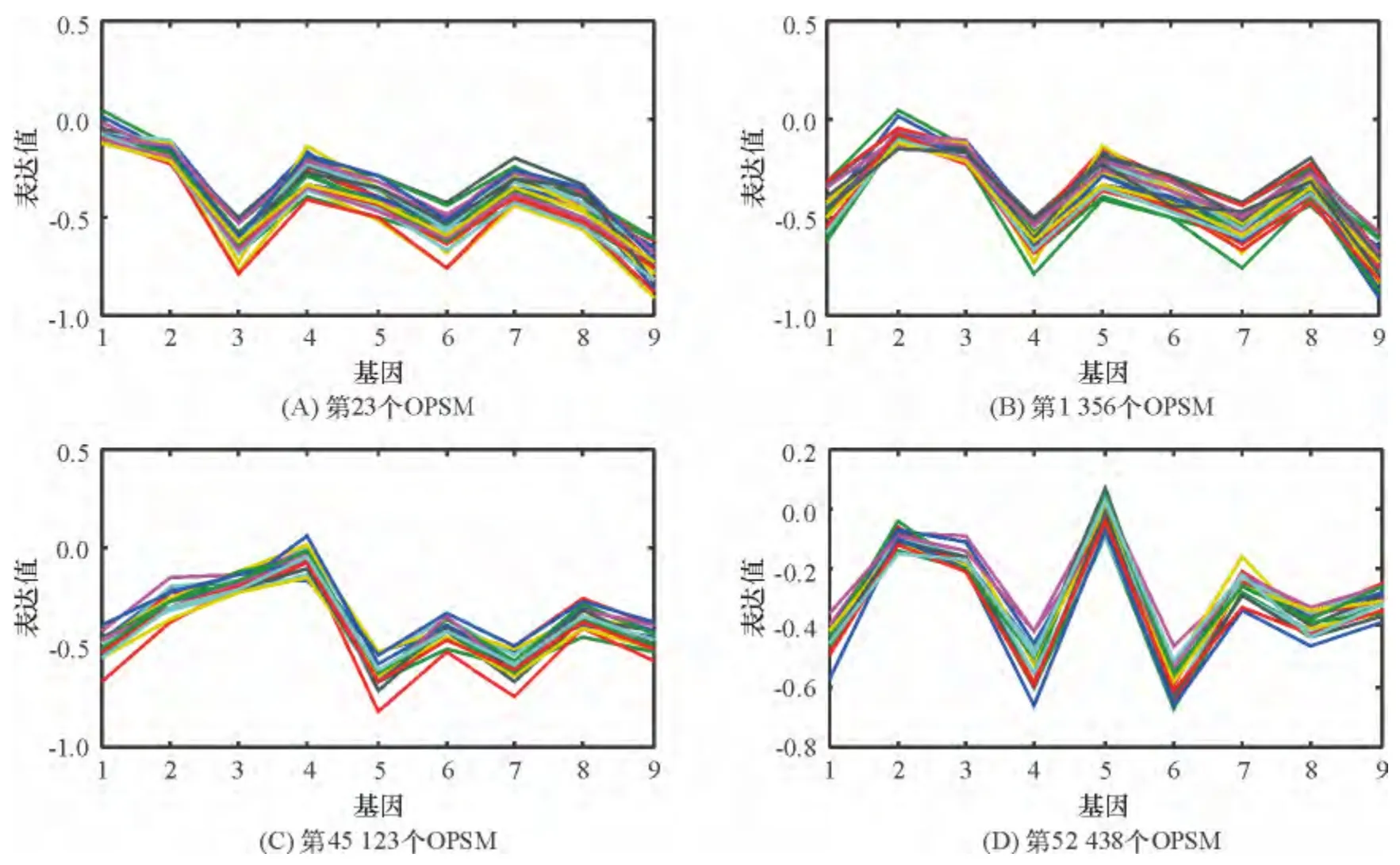
图5 随机挑选实验中找到的4个OPSMs 折线图Figure 5 Broken line charts of OPSMs found in randomly picked experiments
3.2 算法性能比较
对文献[7]算法和本文算法所挖掘出的OPSMs进行可视化操作,分别绘制成折线图进行比较.行阈值均设置为20,本文算法的列阈值设置为6 时,由于剪掉的序列模式较多,所以2 种算法均能在内存负担能承受的范围内挖掘出相应的结果(图6). 均比较第33 790个OPSMs.横坐标为对应OPSMs 聚类的列号,纵坐标为对应的聚类元素在基因表达数据矩阵上的表达值.2 种算法对应结果中每一条折线对应的聚类基因,其表达值在原矩阵中呈现一致的同增同减变化趋势,即为OPSM.
为了挖掘出Deep OPSM 而把2个算法的行阈值降低为2 时,文献[7]算法在运行过程中内存占用量极大,导致结果无法挖掘出来,普通的服务器根本无法承受其内存负担. 本文算法在同等行阈值条件下,运行过程中占用的内存比文献[7]算法占用的内存小,说明本文算法更适合挖掘Deep OPSM.如图7 所示,统计行阈值α 分别为2、3、4 时,可挖掘出列数分别为10 ~15 的Deep OPSM 的数量. 在相同行阈值下,OPSM 数量随着列数的增加而减少;在列数相同时,OPSM 数量随着行阈值的增加而急剧减少. 而行数较少、列数较多的OPSM 往往更具有生物学意义,本算法能有效地将Deep OPSMs 挖掘出来.
3.3 GO 分析
GO(Gene Ontology)[16]注释被用来检验本文算法生成的OPSM 聚类的生物学意义,对产生的聚类结果作真实性验证. GO 是基因本体联合会(Gene Onotology Consortium)所建立的数据库,旨在建立一个适用于各种物种的、对基因和蛋白质功能进行限定和描述的、并能随着研究不断深入而更新的语言词汇标准.
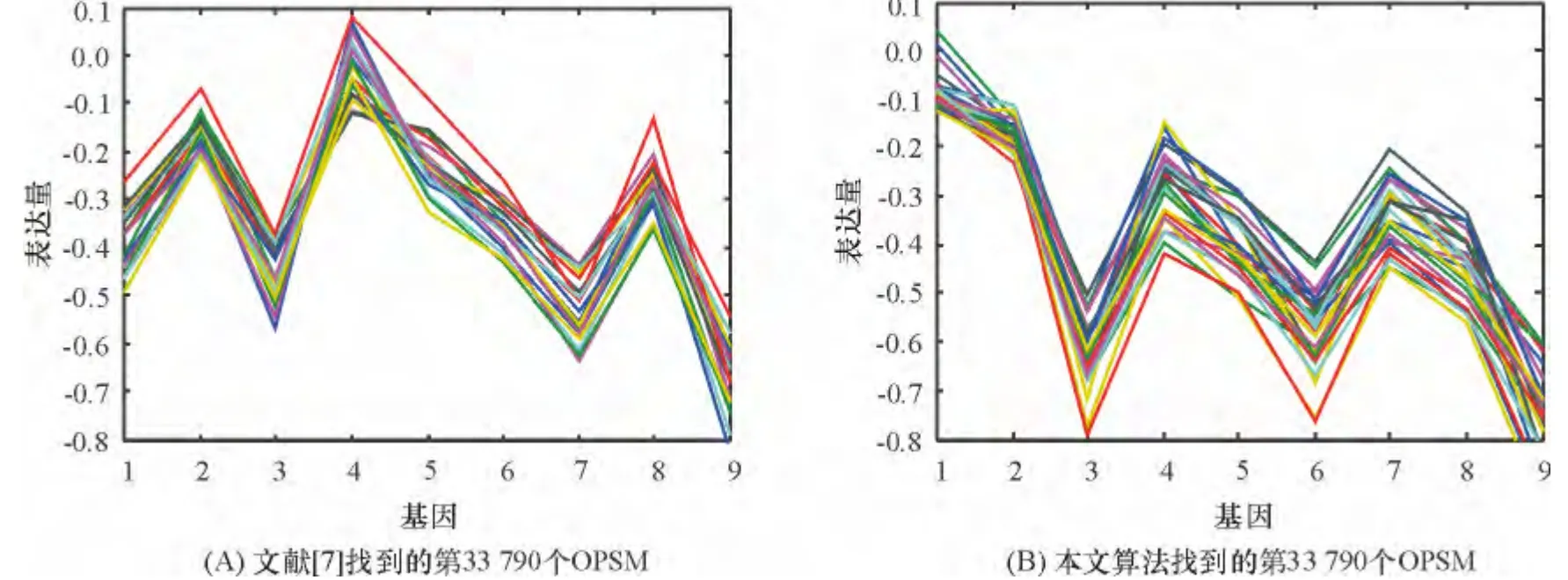
图6 算法性能比较Figure 6 Performance comparisons of two algorithms
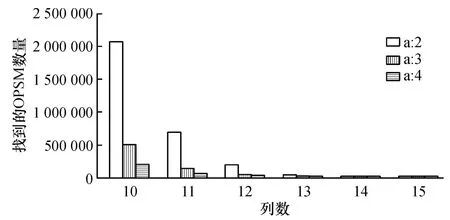
图7 低阈值Deep OPSM 统计图Figure 7 Statistical chart of Deep OPSMs based on low threshold
从本文算法产生的OPSM 聚类中挑选出部分聚类,进行GO 注释实验.结果证明,实验产生的OPSM聚类,能够对应某些基因关联调控的生物特征.列出部分GO 注释的查找结果如表3 所示. 其中当Pvalue 值较低时,说明了该簇基因在1个或者多个GO 项目中具有某些特别的生物学意义. 部分P-value 值小于1.0 ×10-25的GO 项目,表明本文算法能够找到包含关联调控信息的基因簇,从生物意义上证明了本文方法的可行性.
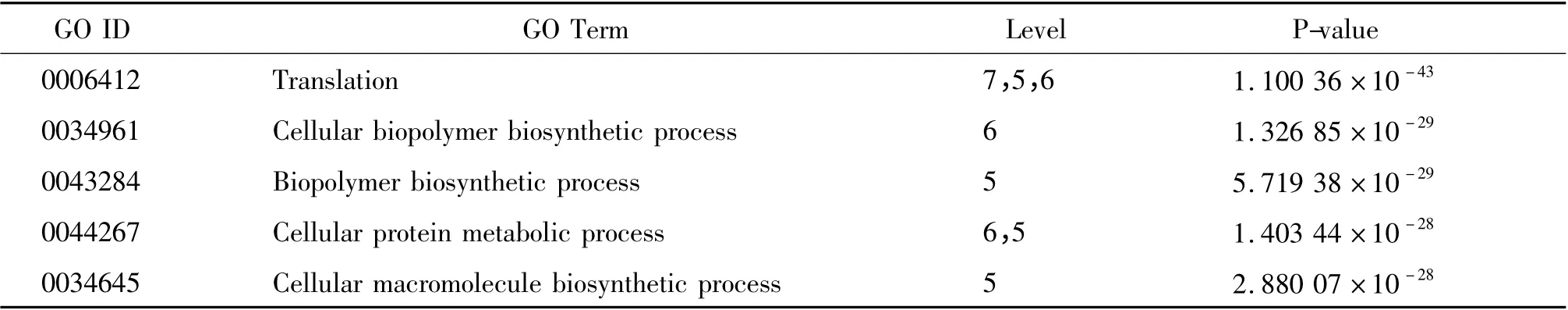
表3 GO 分析部分结果Table 3 Part of GO analysis results
4 结论
OPSM 模型作为一种基于模式的双聚类方法,在分析基因数据矩阵等方面被广泛的应用. 在一个OPSM 聚类中,形成聚类的若干基因在特定的条件子集下有一致的表达模式. 这种关联的共同表达隐含着基因的关联调控,所以在基因数据矩阵上进行的双聚类分析有极大的生物学意义. 本文根据OPSM 模型,建立了一种快速有效的精确性寻找方法,来挖掘分散在基因数据矩阵中的OPSM 聚类.结果证明,该方法能够快速地找到符合条件的OPSM聚类,并且能够通过条件存储,针对长频繁模式进行寻找分析,挖掘出更具生物学意义的Deep OPSM 聚类.此外,通过条件存储,可以在多台计算机上实现并行计算,提高分析处理速度,适应大型数据矩阵的分析需求.最后从生物学(以半乳糖酵母基因数据中的时间序列)的角度,验证了该方法的可行性.
[1]蔡莉,郭红. 一种改进的基因表达数据双聚类算法[J].福州大学学报:自然科学版,2010,38(1):41-47.Cai L,Guo H. An improved biclustering a lgorithm for gene expression data[J]. Journal of Fuzhou University:Natural Science,2010,38(1):41-47.
[2]Lazzeronil L,Owen A. Plaid models for gene expression data[J]. Statistica Sinica,2000,12(1):61-86.
[3]Andrew C T,Prokopyev O A. Solving the order-preserving submatrix problem via integer programming[J]. Informs Journal on Computing,2010,22(3):387-400.
[4]Liu J Z,Wei W. OP-cluster:Clustering by tendency in high dimensional space[C]∥3rd IEEE International Conference on Data Mining. Melbourne,USA,2003:187-194.
[5]Yun X,Li Y T,Deng W J,et al. Mining order-preserving submatrices based on frequent sequential pattern mining[C]∥Health Information Science,HIS 2014.Shenzhen,China,2014:184-193.
[6]Ben-Dor A,Chor B,Karp R,et al. Discovering local structure in gene expression data:The order-preserving submatrix problem[J]. Journal of Computational Biology,2003,10(3/4):373-384.
[7]Cheung L,Kevin Y Y,Cheund D W,et al. On mining micro-array data by order-preserving submatrix[J]. International Journal of Bioinformatics Research A,2007,3(1):42-64.
[8]Fang Q. Effective discovery of order-preserving submatrices[D]. Hong Kong:Hong Kong University of Science and Technology,2012.
[9]Fang Q,Wilfred N G,Feng J L,et al. Mining order-preserving submatrices from probabilistic matrices[J]. ACM Transactions on Database Systems,2014,39(1):1-44.
[10]Hochbaum D S,Levin A. Approximation algorithms for a minimization variant of the order-preserving submatrices and for biclustering problems[J]. ACM Transactions on Algorithms,2013,9(2):1-12
[11]Byron J G,Grifiith O L,Ester M,et al. On the deep order-preserving submatrix problem:A best effort approach[J]. IEEE Transactions on Knowledge and Data Engineering,2012,24(2):309-325.
[12]Williamson M P,Sutcliffe M J. Protein-protein interactions[J]. Biochemical Society Transactions,2010,38(4):875-878.
[13]Albert R. Scale-free networks in cell biology[J]. Journal of Cell Science,2005,118(21):4947-4957.
[14]Ideker T,Thorsson V,Ranish J A,et al. Integrated genomic and proteomic analyses of a systematically perturbed metabolic network[J]. Science,2001,292(5518):929-934.
[15]Yee Y K,Medvedovic M,Bumgarner R E. Clustering gene-expression data with repeated measurements[J].Genome Biology,2003,4(5):1-17.
[16]David M,Brun C,Remy E,et al. GOToolBox:Functional analysis of gene datasets based on gene ontology[J]. Genome Biology,2005,5(12):1-8.
