Study of Press-Induced Phase Transition in PbS
2015-12-14LiWeiHeQinyuWangYinzhen
Li Wei ,He Qinyu,Wang Yinzhen
(Laboratory of Quantum Information Technology,School of Physics and Telecommunication Engineering,South China Normal University,Guangzhou 510006,China)
1 Introduction
The lead sulfide (PbS)is an important narrow band-gap semiconductor with many excellent physical and chemical properties[1-4]. Many of the experimental investigations have been focused on it[5-10]. Despite the progress made in the last few years in the experimental investigations of PbS system,many firstprinciples calculations have been performed to study the electronic and structural properties of PbS system in recent years[11-17]. Most of those were mainly interested in its electronic properties. One of the interesting features of PbS is the strong increase of the band gap energy with increasing temperature or decreasing pressure[18-19]. The pressure effect has been explained as originating from the relatively deep lying Pb s state together with level repulsion around the L point.
Lead sulfide crystallizes in a cubic structure (a phase)under normal pressure. There are two atoms in the unit cell,the lead atom at the center and the sulfide atom at the vertex site. The PbS compounds exhibit structural transitions under pressure. At high pressures,the CsCl structure and several intermediate phases of semiconducting character are found. However,the structural details of those intermediate phases are still debated. Orthorhombic phases of Cmcm symmetry for PbS have recently been reported. Resistivity measurements under high pressure on several lead chalcogenides suggested the existence of a phase transition at 2.2 GPa. The phase transition at 2.2 GPa to a β-phase having orthorhombic symmetry (CrB structure type)was characterized as a first order transition[20]. The mean transition pressure of the cubic structure to a black orthorhombic structure was found to be 3.16 GPa for 10 μm PbS[21]. The experimental transition pressure from the intermediate orthorhombic phase to the CsCl phase is 21.5 GPa for PbS. The study of pressure induced phase transition of α/β-PbS is relatively few,so it was worth reexamining the PbS system under pressure.
The purpose of this paper is to explore the phase transition of α/β-PbS under high pressure. The change of lattice parameters and band structures under pressure are calculated. The transition pressure is obtained through analysis of the relative enthalpy. The phonon properties such as phonon dispersion and phonon density of states (PDOS)are calculated. The effects of phonon softening process on phase transition are checked and discussed.
2 Computational method
The first-principles calculations presented here were performed on the platform of Materials Studio,which was based on density functional theory using a plane-wave basis set for the expansion of the wave functions[22]. Norm-conserving pseudo-potentials were employed throughout the study. Generalized gradient approximation (GGA)was adopted to evaluate exchange-correlation energy. Monkhorst-Pack mesh was used to select 56 k-points for bulk calculation. A plane-wave cutoff energy of 540 eV was employed in the work. It was shown that the results are well converged at this cutoff.
The Pb (5d 6s 6p)and S (3s 3p)were treated as valence state. The geometries for all the systems are optimized. The minimum total energy of the structure is achieved by relaxing the internal coordinates using the Broyden-Fletcher-Goldfarb-Shanno (BFGS)algorithm.Spin-orbit was not involved in the calculations,because the effect of spin-orbit is weak[23]. The following thresholds for converged structures are employed:Energy change per atom less than 2 ×10-6eV,residual force less than 0.1 eV/nm,stress below 0.02 GPa and the displacement of atoms during the geometry optimization less than 0.001 nm.
3 Results and discussions
The lattice parameters and elastic constants of α-PbS under pressure were first calculated,which are shown in Figure 1. In order to study the press-induced structural transition of PbS,we set hydrostatic pressure to PbS solids with two possible types of structural phases (α and β)and fully optimized both the lattice parameters and atomic positions. The bulk modulus (B0)was determined through the Birch-Murnaghan equation. The variations of V/V0,where V was the volume at a given pressure and V0was the volume at p =0 GPa,and the bulk modulus with pressure were shown in Figure 1. A linear decrease was found for V/V0,and a linear increase was found for B0. Here,the bulk modulus at p =0 GPa was in good agreement with the previous calculated value of 55.7 GPa[23]. In this work,the lattice constants were overestimated compared to experiment,resulting in underestimated bulk modulus.
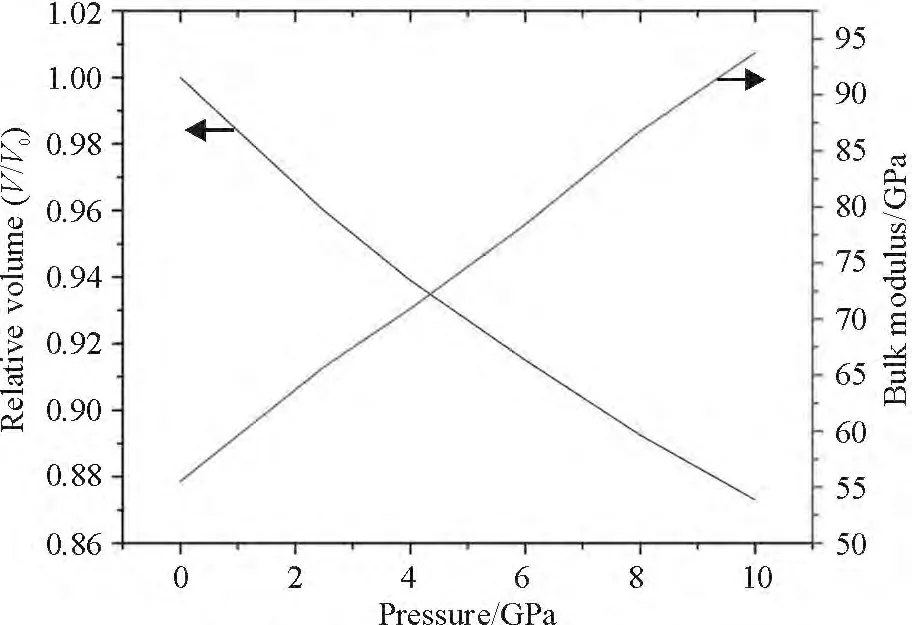
Figure 1 Pressure dependence of the relative volume and bulk modulus for α-PbS
The chemical bond length,atomic charges and bond population are calculated by means of Mulliken analysis in order to understand bonding behavior. From Figure 2 we can find the Pb—S bond is covalent at 0 GPa. However,the covalence of Pb—S bond become weak and the bond length decreases with pressure.
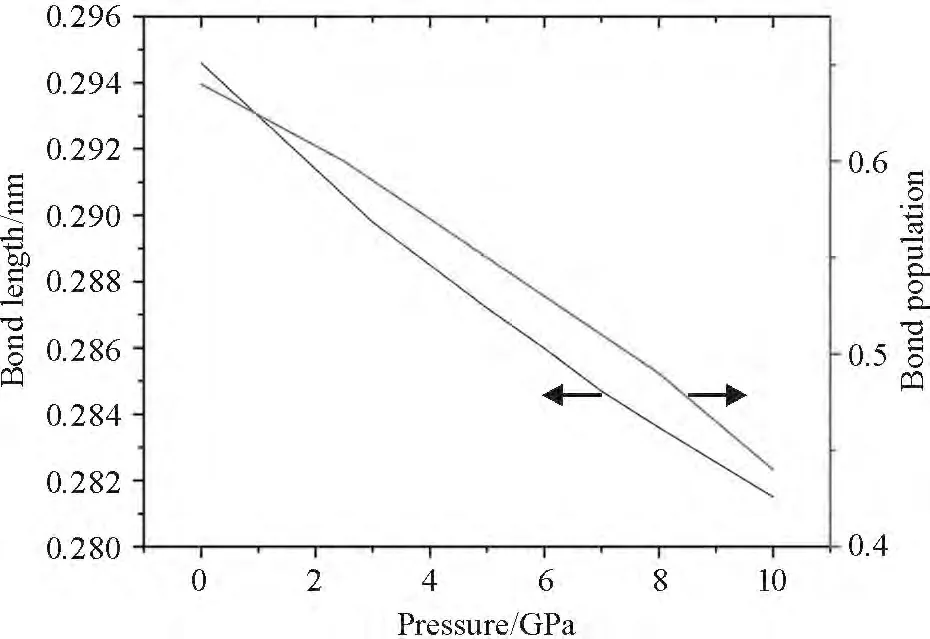
Figure 2 Pressure dependence of bond length and bond population of Pb—S bond in α-PbS
The electronic energy band structures and total electron density of state (DOS)of α-PbS at 0 and 10 GPa are obtained. The calculated band gap of 0.29 eV at 0 GPa is basically consistent with the reports of 0.37 and 0.44 eV[13,23]. Both shapes of electronic energy band structures at 0 and 10 GPa are similar,but showing the negative pressure dependence of the band gap.With pressure,the topmost valence band is shifted upward,and the lowest conduction band is shifted downward in energy. The band dispersion of bands closet to the Fermi level is increased with pressure. There is a significant reduction of the band gap,which is controlled by level repulsions. Because the level repulsion increases when the bond length become shorter,the band gap in α-PbS will decrease with pressure. We think the reason is the conduction band,which has a contribution from the excited 3d orbital of S,is more sensitive to the pressure and eventually crossing the Fermi energy with increasing pressure. Furthermore,we also think the increasing pressure makes the band gap and carrier density actually change,which is probably partly caused by decreasing the bond-length of Pb—S.
The calculated curves of relative enthalpy with pressure for two phases are shown in Figure 3. The transition pressure is the pressure at which the relative enthalpy curves for two phases crosses.
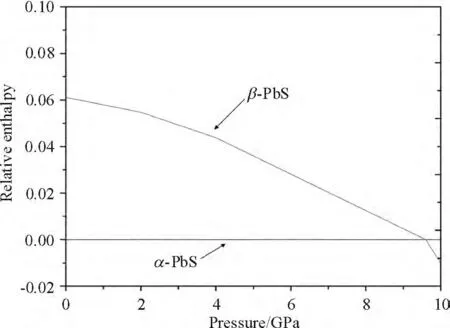
Figure 3 Calculated relative enthalpy versus pressure
From the Figure 3 we find that the calculated transition pressure for the α→β phase transition is about 9.5 GPa. We also can see that the enthalpies of α-PbS and β-PbS are very close around the transition pressure,which suggests that the relative stability of two phases will be sensitive to external factor. So,some fluctuations in the transition pressure maybe occurred with different external factor such as temperature and stress condition. This transition pressure is higher than the experimental value of 2.2 GPa. This is mainly due to the neglect of temperature effects in GGA calculation. Further more,their experiment maybe didn't obtain perfect hydrostatic pressure. So,the discrepancy between calculated and experimental transition pressure can be accepted.
The Born effective charges (BEC)tensor of α-PbS was calculated. The BEC tensor quantifies the macroscopic electric response of a crystal to the internal displacements of its atoms. For α-PbS,the off-diagonal elements of the BEC tensor are all zero and the three diagonal elements Ziare the same,i. e. ZPb=ZS. So,only ZPbis considered here. The increasing BEC suggested that α-PbS could be more easily polarized with distortions or atomic displacements.
To find out whether there exist soft phonon modes,we calculated the phonon properties with pressure. The pressure dependence of vibrational frequencies (dω/dp)is obtained through a linear fitting of ω-p data between 0 and 9.6 GPa,which can be further used to generate the Grüneisen parameter for each mode. The phonon dispersion curves of the α-PbS at 0 and 9.6 GPa are shown in Figure 4. There are obvious splits occurred between the longitudinal-optical (LO)and transverse-optical (TO)at G point,which indicates the large values of BEC. This arises due to the ionic nature of α-PbS. There is no overlap between LA and TO,which suggests weak LA-TO interaction in α-PbS. The dispersions of TO indicated the covalence of Pb—S bond,which corresponds to large value of the Pb—S bond population in Figure 2.
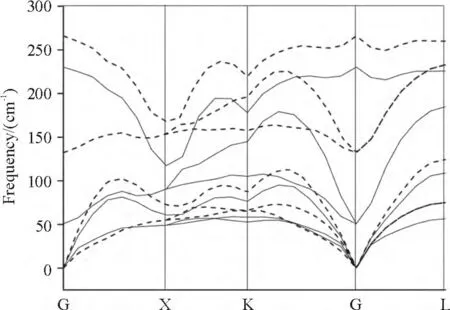
Figure 4 Phonon dispersion curves of α-PbS
As shown in Figure 4,where the solid and dash lines are the phonon dispersion curves at 0 and 9.6 GPa,respectively. We can see that a significant shift of the spectrum toward the high-frequency side is observed at frequencies above 50 cm-1,which indicates large pressure dependences of the high-frequency phonon modes. However,we find there exists a large negative pressure derivative in the transverse-acoustic(TA)mode,which suggests a softening mechanism that can be correlated with atomic displacements in the structure under pressure and lattice instability. A nonlinear dependence of the frequencies on pressure is showed in this soft Mode. TA phonon branches around the G point become soft in the G→X and G→K directions with increasing pressure,which is similar to the reports in[16,24]. These soften phonon branches correspond to Euand B1uvibration modes respectively. So the softening of TA phonons with negative Grüneisen parameter near the zone boundaries appears for α-PbS,which suggests a softening mechanism that can be correlated with atomic displacements in the structure under pressure and lattice instability.
In Figure 5,the PbS4complex 1-2-3-4-5 in the structures is shown. Small balls and big balls denote S and Pb atoms respectively. As shown in Figure 5,there exists a structural unit of PbS4complex in α-PbS,where Pb atom locates on the center of S quadrilateral.There is similar structural unit in β-PbS,but the relative position of Pb atom is out of S quadrilateral. From the calculated phonons DOS of PbS,we know that high-frequency origin mainly contributed by S atoms,and low-frequency origin mainly contributed by Pb atoms. For the PbS4complex in α-PbS,with the softening of B1umode,Pb atom vertically moves out of the plane formed by S atoms along the Z-axis direction. At same time,atoms of PbS4complex changed their sites on x-y plane with the softening of Eu. While the pressure is high enough,the PbS4complex in α-PbS gradually becomes the form of complex in β-PbS. We think this change plays an important role in the transition of α/β phases in PbS.
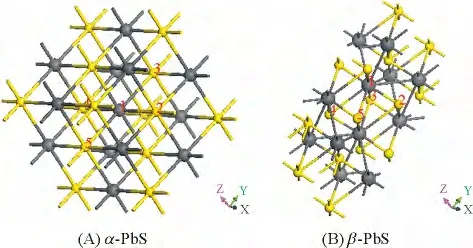
Figure 5 Crystal structures of PbS
4 Conclusions
In present work,we have performed first-principles calculations to study the transition pressure-induced phase transition of PbS. The bond properties,electronic band structures and BEC tensor are calculated using GGA. The transition pressure of 9.6 GPa is obtained through analysis of relative enthalpy with pressure. Softening of phonon modes is usually an important indicator of structural instability,which may cause phase transitions. So the phonon dispersions and DOS of α-PbS were calculated,and we showed that there exist two soft phonon modes (Euand B1u)between 0 and 50 cm-1. These two modes play an important role in understanding the atomic deviations in the α-PbS and the transformation to the β-PbS.
[1]Colvin V L,Schlamp M C,Alivisatos A P. Alivisatos,light-emitting diodes made from cadmium selenide nanocrystals and a semiconducting polymer[J]. Nature,1994,370(6488):354-357.
[2]Ma W,Luther J M,Zheng H,et al. Photovoltaic devices employing ternary PbSxSe1-xnanocrystals[J]. Nano Letter,2009,9(6):2072-2077.
[3]Law M,Beard M C,Choi S,et al. Determining the internal quantum efficiency of pbse nanocrystal solar cells with the aid of an optical model[J]. Nano Letter,2008,8(11):3904-3910.
[4]Plass R,Pelet S,Krueger J,et al. Quantum dot sensitization of organic_inorganic hybrid solar cells[J]. Journal of Physics Chemistry B,2002,106(31):7578-7580.
[5]Saraidarov T,Reisfeld R,Sashchiuk A. Synthesis and characterization of PbS nanorods and nanowires[J].Physica E,2007,37(1):173-177.
[6]Hu K C,Liu P,Ye S J,et al. Ultrasensitive electrochemical detection of DNA based on PbS nanoparticle tags and nanoporous gold electrode[J]. Biosensor and Bioelectron,2009,24(10):3113-3119.
[7]Gaiduk A P,Gaiduk P I,Larsen A N. Chemical bath deposition of PbS nanocrystals:Effect of substrate[J].Thin Solid Films,2008,516(12):3791-3795.
[8]Johnsen S,He J Q,Androulakis J,et al. Nanostructures boost the thermoelectric performance of PbS[J]. Journal of American Chemistry Society,2011,133(10):3460-3470.
[9]Zhao L D,Luo S H,He J Q,et al. High performance thermoelectrics from earth-abundant materials:Enhanced figure of merit in PbS by second phase nanostructures[J]. Journal of American Chemistry Society,2011,133(50):20476-20487.
[10]Zhao L D,He J Q,Wu C,et al. Thermoelectrics with earth abundant elements:High performance p-type PbS nanostructured with SrS and CaS[J]. Journal of American Chemistry Society,2012,134(18):7902-7912.
[11]Wei S H,Zunger A. Electronic and structural anomalies in lead chalcogenides[J]. Physical Review B,1997,55(20):13605-13610.
[12]Tudury G E,Marquezini M V,Ferreira L G,et al.Effect of band anisotropy on electronic structure of PbS,PbSe,and PbTe quantum dots[J]. Physical Review B,2000,62(11):7357-7364.
[13]Hummer K,Grüneis A,Kresse G. Structural and electronic properties of lead chalcogenides from first principles[J]. Physical Review B,2007,75(19):195211.
[14]Zhang L,Song Q,Zhang S B. Exceptionally strong hydrogen bonds affect the surface energy of colloidal nanocrystals:methylamine and water adsorption on PbS[J].Physical Review Letter,2010,104(11):116101.
[15]Romero A H. Experimental studies and ab initio calculations including spin-orbit effects[J]. Physical Review B,2008,78(22):224302.
[16]Kilian O,Allan G,Wirtz L. Near kohn anomalies in the phonon dispersion relations of lead chalcogenides[J].Physical Review B,2009,80(24):245208.
[17]Svane A,Christensen N E,Cardona M,et al. Quasiparticle self-consistent GW calculations for PbS,PbSe,and PbTe:Band structure and pressure coefficients[J].Physical Review B,2010,81(24):245120.
[18]Zasavitskii I I,Silva E,Abramof A E,et al. Optical deformation potentials for PbSe and PbTe[J]. Physical Review B,2004,70(11):115302.
[19]Nimtz G,Slicht B. Springer Tracts in Modern Physics[M]. Berlin:Springer,1983:194-195.
[20]Chattopadhyay T,Schnering H,Grosshans W,et al.High pressure X-ray diffraction study on the structural phase transitions in PbS,PbSe and PbTe with synchrotron radiation[J]. Physica B,1986(139/140):356-360.
[21]Jiang J Z,Gerward L,Secco R,et al. Phase transformation and conductivity in nanocrystal PbS under pressure[J]. Journal of Applied Physics,2000,87(5):2658-2664.
[22]Hohenberg P,Kohn W. Inhomogeneous electron gas[J].Physical Review,1964,136(B864):864-871.
[23]Zhang Y,Ke X Z,Chen C F,et al. Thermodynamic properties of PbTe,PbSe,and PbS:First-principles study[J]. Physical Review B,2009,80(2):024304.
[24]Ei-Sharkawy A A,Ei-Azm A M A,Kenawy M I,et al.Thermophysical properties of polycrystalline PbS,PbSe,and PbTe in the temperature range 300-700 K[J]. International Journal of Thermophysics,1983,4(3):261-269.
