吲哚喹唑啉衍生物抗癌活性与拓扑参数的定量关系
2015-12-11李剑堵锡华
李剑+堵锡华
通讯作者,Email:12dxh@sina.com,唐赛杰2(1.徐州开达精细化工有限公司,中国 徐州221009; 2 徐州工程学院化学化工学院,中国 徐州221111)
摘要为建立吲哚喹唑啉衍生物类药物抗癌活性的定量结构活性相关性模型,分析了20个具有不同取代基的吲哚喹唑啉衍生物分子抗癌活性与分子连接性指数mX及其电性拓扑状态指数Im的关系,有效地表征了该衍生物的分子结构.采用多元线性逐步回归方法进行多次优化筛选了2种分子连接性指数0χp,2χp和2种电性拓扑状态指数I7,I16,经逐步回归分析得到了用于预测吲哚喹啉衍生物抗癌活性的定量结构活性相关(QSAR)模型,回归方程的相关系数为0.820.利用方程计算得到抗癌活性的估算值与实验值之间平均误差只有0.010,此外对模型稳定性和预测能力进行了检验,结果表明模型具有良好的稳健性和预测能力.
关键词拓扑参数;吲哚喹唑啉衍生物;抗癌活性;定量结构活性相关性
中图分类号O641文献标识码A文章编号10002537(2015)06004604
Relationship Between Anticancer Activities of Indolo [1,2b]
Quinazoline Derivatives and Topology Parameters
LI Jian1, DU Xihua2*, TANG Saijie2
(1.Xuzhou Kaida Fine Chemical Co., Ltd.,Xuzhou 221009, China;
2.School of Chemistry and Chemical Engineering, Xuzhou Institute of Technology, Xuzhou 221111, China)
AbstractIn order to establish QSAR model of the antitumor activity of indolo[1,2b] quinazoline derivatives, the relationships amongst the molecular connectivity indices mX, the electrotopological state indices Im and the anticancer activities of 20 indolo[1,2b] quinazoline derivatives were analyzed, which nicely represent the molecular structure of the derivatives. Molecular connectivity indices 0χp, 2χp and electrotopological state indices I7, I16 were selected by multiple linear stepwise regression method. The QSAR model was established by stepwise regression analysis and the correlation coefficient is 0.820. The average error between estimated value and experimental value is 0.010. Moreover, the examination of the stability and prediction ability of the model in this paper shows that the model has good stability and prediction ability.
Key wordstopology parameters; indolo[1,2b]quinazoline derivatives; anticancer activity;QSAR
结肠癌是最常见的恶性肿瘤之一,被认为是癌症引发死亡的第三大杀手.近年来我国结肠癌的发病率也呈逐年上升趋势[12].目前,治疗结肠癌的方法并不多,治疗结肠癌的药物一般用氟尿嘧啶类药物,如5氟尿嘧啶和亚叶酸等,但该类药物不良反应严重,毒副作用大.吲哚喹唑啉是一种可从多种医用植物中提取的生物碱,具有良好的抗肿瘤、抗菌作用以及低的细胞毒性,特别是对结肠癌有良好的治疗作用.
定量结构活性相关性研究(QSAR)是以结构为基础的一种定量药物设计的理论方法;它涵盖了计算机科学、化学、统计学、毒理学以及生物学的一些内容,该法能揭示分子物化性质和生物活性/毒性与分子结构信息参数之间的变化规律,被广泛运用于许多领域,如合成化学、环境毒性、农药研制、生物分子模拟以及药物设计与评价等等.将QSAR方法应用于吲哚喹唑啉衍生物研究,可为分子设计和药物活性(毒性)评价起到一个重要的作用.本文通过研究分子连接性指数[34]和电性拓扑状态指数与文献[1]吲哚喹唑啉衍生物抗癌活性之间的关系,构建了定量结构活性相关模型,获得较为满意的效果,本法对高活性抗癌性吲哚喹唑啉类衍生物药物的研究、设计和开发利用具有较好的实际意义.
QSAR方法是根据化合物分子和生物体内受体分子的立体结构特征为基础[5]、根据物质分子间的相互作用能量来探讨化合物分子的结构模型与其生物体性质之间存在的定量关系.本文首先利用软件Chemoffice 2005建构吲哚喹唑啉衍生物的结构图,再通过MATLAB自编程序软件[6]计算得到吲哚喹唑啉衍生物的分子连接性指数、分子形状指数、分子电性拓扑状态指数数值,用SPSS统计软件经过多次优化筛选参数,最后选择得到分子连接性指数mX和电性拓扑状态指数Im,经过回归分析,建立了相关性较好的定量构效关系模型.
1指数的构建
为构建最佳的预测模型,参数的选择最为重要,它既要求参数与性质之间有良好的相关性,又要求据此所建模型有良好的稳健性和预测能力,而且还要求样本数与参数个数的比值应大于5,基于这些基本原则,本文利用相关的程序软件计算得到了相关参数并进行了筛选.
湖南师范大学自然科学学报第38卷第6期李剑等:吲哚喹唑啉衍生物抗癌活性与拓扑参数的定量关系1.1分子连接性指数(mX)
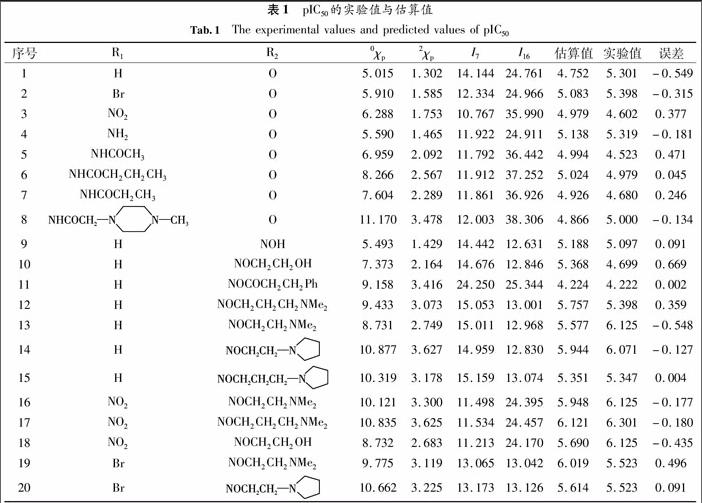
分子连接性指数能够反应分子中原子的连接排列方式,它是一种主要反应分子结构信息的参数,1975年由Randic[7]在研究小分子烷烃的数目与沸点关系时提出的分支指数,由于该指数只适用于烃类化合物,后来Kier等人[8]对其进一步修正,在分子隐氢图的邻接矩阵基础上提出一种新的结构量化数值,它具有广泛的应用性和准确性,它不仅能够处理杂原子和不饱和键,还能处理环和芳香类复杂分子.它在药物分子设计和合成方面显示出了重要的作用[9],但是分子连接性指数同时也存在着一定的局限性[10],利用其单一指数与药物性质进行关联,相关性并不是很理想,还需要与其他指数结合使用才有效果.本文通过程序计算了文献[1]中20个化合物分子的分子连接性指数,再通过回归分析优化筛选,确定了其中二种连接性指数0χp、2χp作为与药物分子抗癌活性关联的变量值,相关数据见表1.
1.2电性拓扑状态指数(Im)
电性拓扑状态指数(Im)是一种结构参数,它是根据在分子中的成键电子信息与拓扑环境来表征分子中每种非氢原子类型,由Hall和Kier等人[11]提出,主要针对连接性指数的局限性提出的一种新的概念指数,本指数充分考虑了电性特征[12],它是在原子水平上的拓扑指数,包含分子特征结构的电子信息和拓扑信息,能反映原子所受分子环境的影响,目前国内将电性拓扑状态指数应用于药物抗癌活性方面的研究报道并不多见,因此利用本指数来研究吲哚喹唑啉衍生物的原子结构对分子性质的影响有实际意义.
在前面工作[1315]基础上,本文通过程序计算了文献[1]中的20个化合物分子的电性拓扑状态指数,再通过回归分析优化筛选,确定了其中二种电性拓扑状态指数I7和I16作为方程变量,相关数据见表1.
2QSAR模型的建立
分析文献[1]中20种吲哚喹唑啉化合物,运用SPSS统计分析软件对2个分子连接性指数和2个电性拓扑状态指数与抗癌活性值进行逐步回归分析,建立的线性回归方程为
pIC50= 9.444-0.6710χp+2.039 2χp-0.201 I7-0.046 I16,(1)
N=20,R=0.820,R2=0.673,S=0.390,F=7.723.
其中 pIC50为结肠癌细胞半抑制浓度的负对数,N 为样本数,R为相关系数,R2为判定系数,S为估计标准误差,F为Fischer 检验值.可以看出方程的相关系数达到了0.820,这对于药物类的QSAR模型来说,达到了较好的相关性.根据模型得到的pIC50的估算值与文献实验值所得的数值吻合度较好,两者的平均绝对误差为0.010,相关数据见表1.
与文献[1]相比,虽然本法的标准偏差较高,但本法使用自编程序计算简便、方法简单,具有一定的实用性;而文献[2]的标准偏差虽然很低,但该文为17个样本分子,本文中的3,10,11,12号分子没有列入该文中(另加一分子),从这4个分子的pIC50实验值可以看出,它们明显属于离群数据,将它们与其他分子放在一起进行回归分析会导致偏差增大.
表1pIC50的实验值与估算值
Tab.1The experimental values and predicted values of pIC50
序号R1R20χp2χpI7I16估算值实验值误差1HO5.0151.30214.14424.7614.7525.301-0.5492BrO5.9101.58512.33424.9665.0835.398-0.3153NO2O6.2881.75310.76735.9904.9794.6020.3774NH2O5.5901.46511.92224.9115.1385.319-0.1815NHCOCH3O6.9592.09211.79236.4424.9944.5230.4716NHCOCH2CH2CH3O8.2662.56711.91237.2525.0244.9790.0457NHCOCH2CH3O7.6042.28911.86136.9264.9264.6800.2468O11.1703.47812.00338.3064.8665.000-0.1349HNOH5.4931.42914.44212.6315.1885.0970.09110HNOCH2CH2OH7.3732.16414.67612.8465.3684.6990.66911HNOCOCH2CH2Ph9.1583.41624.25025.3444.2244.2220.00212HNOCH2CH2CH2NMe29.4333.07315.05313.0015.7575.3980.35913HNOCH2CH2NMe28.7312.74915.01112.9685.5776.125-0.54814H 10.8773.62714.95912.8305.9446.071-0.12715H 10.3193.17815.15913.0745.3515.3470.00416NO2NOCH2CH2NMe210.1213.30011.49824.3955.9486.125-0.17717NO2NOCH2CH2CH2NMe210.8353.62511.53424.4576.1216.301-0.18018NO2NOCH2CH2OH8.7322.68311.21324.1705.6906.125-0.43519BrNOCH2CH2NMe29.7753.11913.06513.0426.0195.5230.49620Br 10.6623.22513.17313.1265.6145.5230.0913QSAR模型稳定性和预测能力的检验
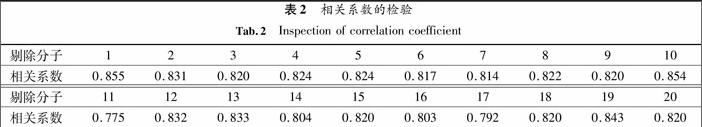
为了检验结构模型(即QSAR模型)中是否存在异常值,用Jackknife法对模型进行稳定性检验,每次剔除一个化合物, 利用剩余的数据进行建模, 得到一个新的相关系数值,重复20次,得到20个相关系数值,大部分相关系数的检测都分布在0.820附近,0.775~0.800之间的有2个,其他18个相关系数都是在0.800~0.855之间波动,具有良好的正态分布,只有剔除1号分子模型的相关系数是0.855,相关系数的平均值与模型的相关系数R是一致的;说明模型的稳定性较好.将这些相关系数列入表2.
采用MINITAB软件的最佳变量子集回归方法对其进行变量的提取和回归统计分析, 可以看出,如使用2个变量,方程最好的R2=0.445,S=0.478;如使用3个变量,方程最好的R2=0.599,S=0.418;而使用4个变量,则是本文的R2=0.673,S=0.390;此时交互检验的相关系数也最好,所以本文采用4个变量比较合理.
表2相关系数的检验
Tab.2Inspection of correlation coefficient
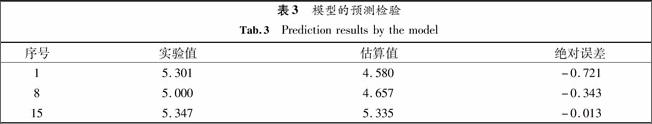
剔除分子12345678910相关系数0.8550.8310.8200.8240.8240.8170.8140.8220.8200.854剔除分子11121314151617181920相关系数0.7750.8320.8330.8040.8200.8030.7920.8200.8430.820为了检验模型的预测能力,随机在剔除1,8,15号分子时得到的相应定量结构相关模型对1,8,15号分子的抗癌活性值进行预测,得到相关的值见表3.
表3模型的预测检验
Tab.3Prediction results by the model
序号实验值估算值绝对误差15.3014.580-0.72185.0004.657-0.343155.3475.335-0.013通过表3可见,模型的实验值与预测值基本吻合,几个数据的平均误差为-0.359,说明模型具有一定的预测能力.
4结果与讨论
本文运用分子连接性指数mX和电性拓扑状态指数Im表征吲哚喹唑啉衍生物的分子模型结构,预测其抗癌活性,两者之间建立较好的定量结构活性相关研究模型,由方程可知,文献[1]中20种吲哚喹唑啉衍生物的pIC50与电性拓扑状态指数呈现正相关,通过对估算值与实验值的比较可见,当引入的取代基团R含有杂环时,实验值与利用方程得到的估算值比较吻合;随着引入的R1基团的拉电子能力增加,同时取代基R2的大小相对适中时,模型对预测该类化合物的抗癌活性具有很好的效果;当取代基R1相同时,该类化合物的抗癌活性与取代基R2基团的大小有较大关系,取代基R2的空间效应对化合物影响大;当取代基R2相同时,取代基R1的电性对化合物抗癌活性影响较大,其拉电子能力越强,化合物抗癌活性越好.
综上所述,本文运用分子连接性指数mX和电性拓扑状态指数Im预测吲哚喹唑啉衍生物药物的抗癌活性,建立了较好的定量结构活性相关研究模型,通过Jackknife法检验,模型有较好的稳健性和预测能力.因此运用本法研究吲哚喹唑啉衍生物药物,可以在理论上设计出抗癌活性较高的新型吲哚喹唑啉衍生物,为实验工作者合成活性强的新型药物提供理论依据.
参考文献:
[1]吴文娟,赖瑢,郑康成,等. 抗癌性吲哚喹唑啉衍生物的定量构效关系[J]. 物理化学学报, 2005,21(1):2832.
[2]钱力,沈勇,陈锦灿,等. 抗癌性吲哚喹唑啉衍生物3DQSAR研究及其分子设计[J]. 物理化学学报, 2006,22(11):13721376.
[3]WIENER H. Structural determination of paraffin boiling points [J]. J Am Chem Soc, 1947,69(1):1720.
[4]李鸣建, 冯长君. 取代苯甲酸对植物生长调节活性的拓扑QSAR研究[J]. 哈尔滨工业大学学报, 2009,41(5):195197.
[5]冯长君,杨伟华,沐来龙,等. N,N二甲基2溴苯乙胺类衍生物对大鼠生物活性的三维构效关系研究[J]. 化学学报, 2006,64(12):12131217.
[6]胡黔楠, 梁逸曾, 王亚丽, 等. 直观队列命名法的基本原理及其在矩阵与拓扑指数计算中的应用[J]. 计算机与应用化学, 2003,20(4):386390.
[7]RANDIC M. Characterization of molecular branching[J]. J Am Chem Soc, 1975,97(23):66096615.
[8]KIER L B, HALL L H. Molecular connectivity in structure activity analysis[M]. England: Research Studies Press, 1986.
[9]堵锡华,陈艳,魏家荣. 胸腺嘧啶类抗艾滋药物的构效关系[J]. 武汉大学学报:理学版, 2013,59(1):6670.
[10]SOSKI C M, PLAVSIC D. QSAR study of 1,8Naphthyridin4ones as inhibitors of photosystemII[J]. J Chem Inf Model, 2005,45(4):930938.
[11]HALL L H, KIER L B. Electrotopological state indices for atom types: a novel combination of electronic,to pological, and valence state information [J]. J Chem Inf Comput Sci, 1995,35(6):10391045.
[12]冯长君,唐自强,杨伟华,等. 用分子形状指数、电拓扑态指数研究酚类化合物的生物降解性[J]. 武汉科技大学学报:自然科学版, 2007,30(2):156160.
[13]堵锡华. 多溴代二苯并呋喃/噻吩热力学性质的定量构效关系[J].化工学报, 2010,61(12):30593066.
[14]陈艳,堵锡华. 多氯代二苯并呋喃Ah受体结合能力的QSAR研究[J]. 湖南师范大学自然科学学报, 2009,32(2):7275.
[15]堵锡华.多氯联苯热力学性质的构效关系[J]. 化工学报, 2007,58(10):24322436.
