稳定性同位素稀释-超高效液相色谱串联质谱法测定腐竹中的乌洛托品
2015-08-02张晓鸣彭飞进
徐 幸,张晓鸣,张 燕,舒 平,彭飞进
(1.江南大学食品学院,江苏无锡 214122;2.大理州质量技术监督综合检测中心,云南大理 671000)
稳定性同位素稀释-超高效液相色谱串联质谱法测定腐竹中的乌洛托品
徐 幸1,2,张晓鸣1,*,张 燕2,舒 平2,彭飞进2
(1.江南大学食品学院,江苏无锡 214122;2.大理州质量技术监督综合检测中心,云南大理 671000)
采用超高效液相色谱-串联质谱法(UPLC-MS/MS)和稳定性同位素稀释技术,建立了测定腐竹中乌洛托品的分析方法。以乙腈为提取溶剂,向腐竹样品中加入乌洛托品的稳定性同位素,经固相萃取柱净化,采用HILIC色谱柱分离,目标物在UPLC-MS/MS的多反应监测(MRM)模式下,内标法定量。该方法在1~40 μg/L范围内有良好的线性关系,相关系数为0.9996,方法定量限为2 μg/kg。在添加水平为2、10、30 μg/kg时,平均回收率为97.8%~101.5%,相对标准偏差(RSD)为1.9%~4.8%。本方法灵敏度高,准确度和重复性好,可为作为检测腐竹中违法添加乌洛托品的方法。
稳定性同位素稀释技术,超高效液相色谱-串联质谱法,乌洛托品,腐竹
乌洛托品(Urotropine),化学名称为1,3,5,7-四氮杂三环[3.3.1.1]癸烷,分子式为C6H12N4,常用于医药和化工领域[1],因其在酸性环境中会分解为甲醛而具有杀菌作用,常被用作防腐剂非法添加到食品中。因甲醛对人体的免疫功能以及消化系统有损害[2],早在2010年3月,中国就将乌洛托品列入食品中可能违法添加的非食用物质名单(第四批)[3]。但有监管部门已发现,部分生产者将乌洛托品非法添加到腐竹中,以延长其保质期。然而我国尚无检测腐竹中乌洛托品的国家标准,仅有只适用于检测动物源性食品的行业标准SN/T 2226-2008[4]。近年来,关于豆制品中乌洛托品的检测方法已有文献报道,主要有气相色谱法[5]、液相色谱法[6]、激光拉曼光谱法[7]以及液质联用法等[8-10],绝大多数都采用外标法来定量,而外标法的回收率不稳定,相对标准偏差较大,这将导致检测数据的不准确。也有研究者用内标方法来定量,陆春良等[11]采用三聚氰胺作为内标对乌洛托品进行定量检测,但两者的化学性质并非完全一致,这也会使检测数据不准确。因此,有必要建立一种更稳定可靠的检测方法。
稳定性同位素稀释技术用于食品检测中,不仅能消除基质效应的影响,还可以使定量更准确[12-13]。本实验采用同位素标记的乌洛托品作为内标,超高效液相色谱-串联质谱法测定乌洛托品的含量,使得检测方法的精密度和准确度都得到显著提高。
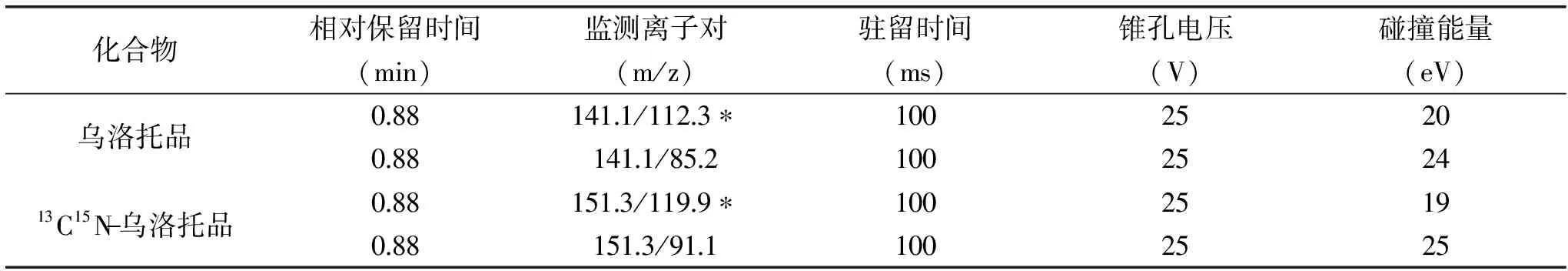
表2 质谱分析参数
注:
注:*标注的为定量离子对。
1 材料与方法
1.1 材料与仪器
乌洛托品、13C15N-乌洛托品标准品 购于Sigma公司,纯度≥99%;色谱纯甲醇、乙腈 购于Merck公司;乙酸铵 色谱纯,购于Acros公司;所用其他试剂 均为分析纯;实验室用水 超纯水;腐竹样品 购于当地市场。
Cleanert PCX固相萃取小柱 规格为60 mg/3 mL,购于Agela Technologies公司;0.22 μm尼龙滤膜 购于安谱科学仪器有限公司;QTRAP 4500串联质谱联用仪、Ekspert ultraLC 100-XL超高效液相色谱仪 购于AB SCIEX公司;色谱柱为Phenomenex Luna 3μ HILIC,规格为100×2.0 mm,3μm;氮吹仪;超声波清洗仪;AL204电子天平 购于Mettler toledo公司。
1.2 色谱-质谱分析条件
液相色谱操作条件:流速:0.6 mL/min;柱温:40 ℃;进样体积:5 μL;流动相:A为0.1%甲酸-5 mmol/L乙酸铵溶液,B为乙腈,梯度洗脱条件如表1。
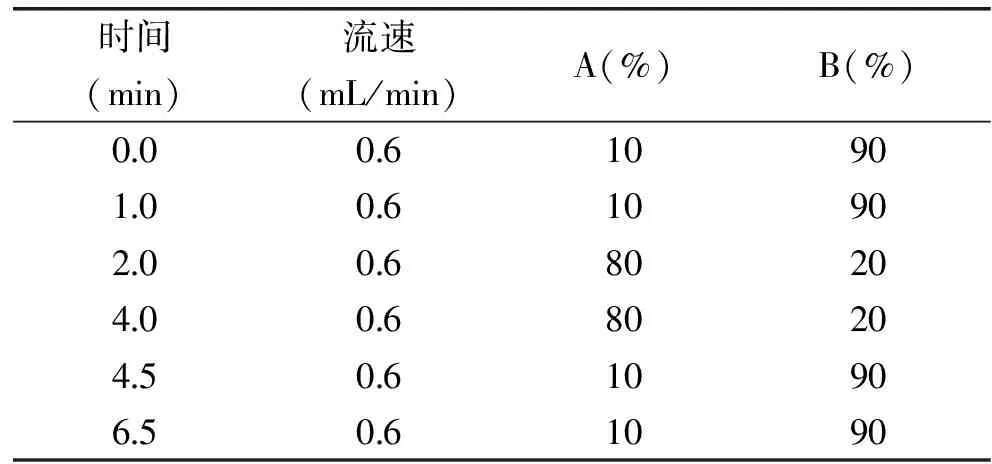
表1 流动相的梯度洗脱程序
质谱离子源选用电喷雾正离子扫描模式(ESI+);多反应监测(MRM);离子源温度为110 ℃;喷雾电压为5500 eV;脱溶剂温度为450 ℃;质谱分析参考条件参见表2。
1.3 标准溶液的配制和曲线绘制
准确称取50 mg乌洛托品标准品与13C15N-乌洛托品标准品,用乙腈溶解并分别定容至50 mL,配制成1 mg/mL的标准物质储备溶液,避光保存于4 ℃冰箱中。再逐级稀释配制成标准工作液,将乌洛托品配制成1、2、5、10、20、30、40 μg/L的标准系列,13C15N-乌洛托品在每个浓度点的浓度都为10 μg/L,绘制标准曲线。
1.4 样品前处理
1.4.1 样品的制备与提取 取有代表性样品约100 g,用组织捣碎机捣碎,准确称取2.5 g试样(精确到0.001 g)于50 mL比色管中,加入100 μg/L的13C15N-乌洛托品标准溶液250 μL,用乙腈定容至25 mL,超声提取15 min,用快速定性滤纸过滤,吸取10 mL滤液于40 ℃下氮吹浓缩近干后待净化。
1.4.2 净化 固相萃取柱使用前,先依次用3 mL甲醇、3 mL水活化,用2 mL甲醇溶解浓缩后的残渣,并过固相萃取柱,再用3 mL水和3 mL甲醇淋洗固相萃取柱,弃去流出液,在15 mmHg以下减压抽至柱体干涸,用5 mL 5%氨水-甲醇(v/v)洗脱并收集洗脱液,于40 ℃下氮气吹干,准确加入1 mL乙腈溶解残渣,经0.22 μm尼龙滤膜过滤,供液相色谱串联质谱分析测定。
2 结果与分析
2.1 质谱条件的选择
采用针泵直接进样的方式,分别对乌洛托品与13C15N-乌洛托品标准溶液进行母离子扫描,以确定目标物的准分子离子,两种物质在正离子模式下灵敏度都比负离子模式下响应高。再分别以准分子离子m/z 141.1与m/z 151.3作为母离子,对其子离子进行全扫描,获得了乌洛托品与13C15N-乌洛托品的质谱图,如图1、图2所示,由质谱图可以看出乌洛托品与13C15N-乌洛托品的特征离子,选择其中响应较高,干扰较小的离子对作为多反应监测的离子对,SN/T2226-2008推荐的参考离子对为m/z 141.1/98.2,而实际检测时发现尽管此离子对的响应较高,但样品中的干扰也较大,故最终选择了干扰较小的m/z 141.1/85.2作为参考离子对。在选定多反应监测的离子对后,对仪器的锥孔电压、驻留时间以及碰撞能量等参数进行优化选择,优化后的参数见表2。
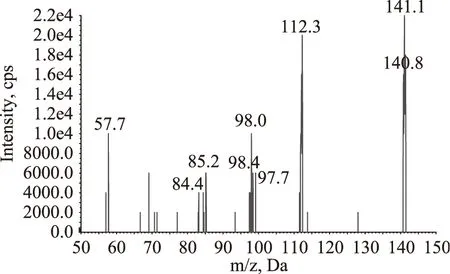
图1 乌洛托品的全扫描质谱图Fig.1 Full scan mass spectrum of hexamethylenetetramine
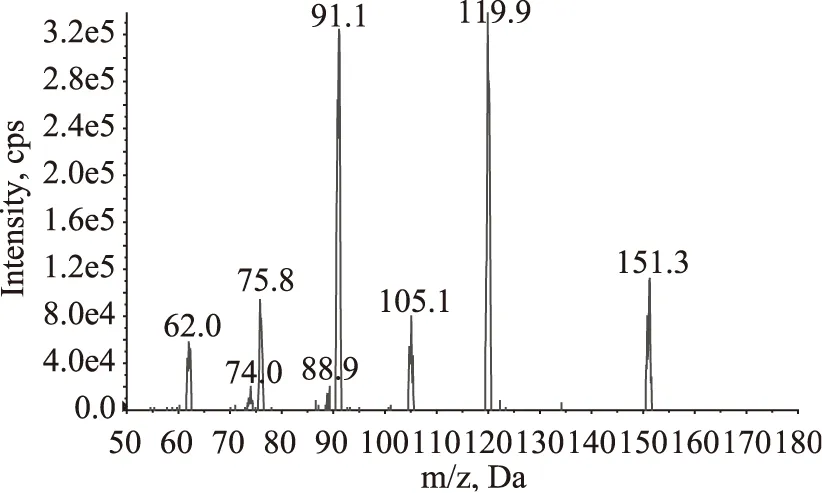
图2 13C15N-乌洛托品的全扫描质谱图Fig.2 Full scan mass spectrum of13C15N-hexamethylenetetramine
2.2 色谱条件的优化
实验考察了不同流动相对乌洛托品与13C15N-乌洛托品响应的影响,包括水-乙腈、5 mmol/L乙酸铵溶液-乙腈、0.1%甲酸-5 mmol/L乙酸铵溶液-乙腈等体系对质谱响应的影响,结果显示采用0.1%甲酸-5 mmol/L乙酸铵溶液-乙腈体系时乌洛托品与13C15N-乌洛托品的响应最强,因此,最终选择0.1%甲酸-5 mmol/L乙酸铵溶液-乙腈体系作为流动相,并对流动相的梯度洗脱程序进行了优化,优化后的条件见表1,图3为乌洛托品与13C15N-乌洛托品在此条件下的多反应监测图。
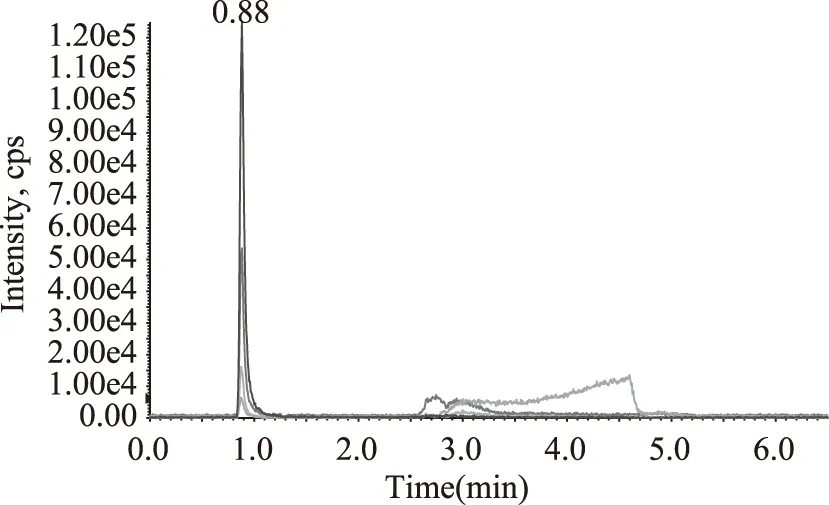
图3 多反应监测色谱图Fig.3 Multiple reaction monitoring chromatogram
2.3 样品前处理条件的选择
乌洛托品溶于水、乙腈、甲醇、乙醇等极性溶剂。因腐竹中含有大量蛋白质,选择提取溶剂时,需兼顾沉淀蛋白与提取目标物的作用。有文献报道[14-15]采用高氯酸或三氯乙酸的水溶液提取豆制品中的乌洛托品,但乌洛托品在酸性条件下易分解[3],因此本实验选用非酸性溶剂乙腈、甲醇和乙醇等来提取乌洛托品,用乙醇提取时,乌洛托品的回收率低于50%,用乙腈和甲醇提取时回收率可达60%~70%,而乙腈提取的滤液色素含量较甲醇提取的低,最终采用了乙腈作为提取溶剂。
乌洛托品分子属于多环叔胺结构,溶液呈碱性,在溶液中离子带正电荷,因此需选择能与目标物发生离子交换的阳离子交换柱,已有文献报道混合型阳离子交换柱的回收率,较其他类型的阳离子交换柱好[4,8,14-15],也有文献报道,采用PXA阴离子交换小柱作为净化柱用于乌洛托品的检测[3,9],但阴离子交换柱的净化原理是杂质吸附于柱子中,目标物直接随溶剂流出,如样品中含有大量盐类,不能保证收集的流出液中不含盐类,此净化效果将不及阳离子交换柱。结合乌洛托品的化学性质,参考文献的报道,本实验采用混合型阳离子交换固相萃取柱作为净化柱。
2.4 方法的线性范围与检出限
乌洛托品的标准系列在浓度范围为1~40 μg/L时,校准曲线的回归方程为Y=0.1049X+0.0164(其中Y为乌洛托品对13C15N-乌洛托品的峰面积比,X为乌洛托品的浓度),线性相关系数为0.9996。根据10倍信噪比(S/N)计算该方法的定量限为2 μg/kg。同时考察了添加在腐竹中的乌洛托品MRM色谱图,在目标物保留时间附近,无明显干扰物存在。
2.5 方法回收率与精密度
分别向腐竹样品中添加浓度为2、10和30 μg/kg三水平的回收率实验,每个浓度重复测定6次,结果见表3,采用同位素内标法定量,其平均回收率在97.8%~101.5%范围,相对标准偏差均小于5%,方法的准确度和精密度都符合分析要求,根据已有的文献数据,采用液相色谱串联质谱-外标法测定食品中的乌洛托品,汪辉等[8]测得的平均回收率在52.6%~118%范围,相对标准偏差为7.1%~10.2%,马雪涛等[14]测得的平均回收率在76.0%~83.6%范围,相对标准偏差为2.7%~5.8%,回收率与内标法比较偏低,且精密度和稳定性也没有内标法好,因此采用同位素内标法使得检测结果更准确。此结果表明乌洛托品的绝对回收率难以达到100%,而稳定性同位素13C15N-乌洛托品与其化学性质类似,采用相同的提取净化过程,两者的绝对回收率较接近,当采用两者的比值作为相对回收率时,精密度与准确度就得到显著提高。
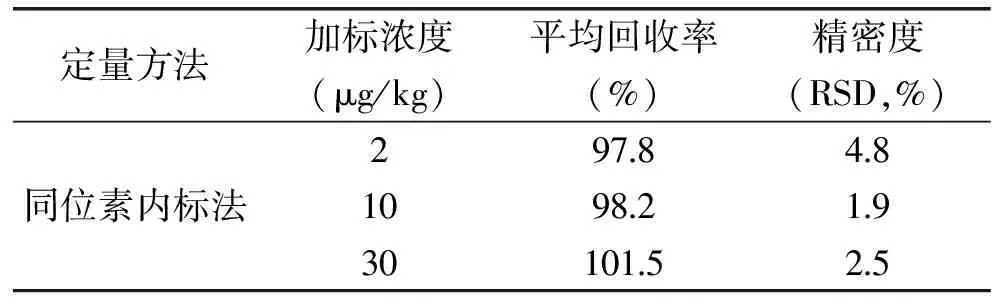
表3 乌洛托品在腐竹中的回收率和精密度实验(n=6)
3 结论
本文建立了针对腐竹样品中乌洛托品的检测方法,采用乙腈提取腐竹中的乌洛托品,同时沉淀了基体中的蛋白,采用乌洛托品的稳定性同位素标记物作为内标,与常用的外标法相比较,同位素稀释法的相对回收率较高,定量检测结果也更准确。采用此法检测了当地市场上的20个腐竹样品,并未检出乌洛托品。此方法操作简便,准确度和精密度较外标法更好,可用于日常食品监管部门对腐竹中乌洛托品的监测。
[1]冼燕萍,陈立伟,罗东辉,等. UPLC-MS/MS测定腐竹和米粉中的乌洛托品[J]. 江南大学学报:自然科学版,2012,11(1):78-82.
[2]吴颖,张慧,崔芳,等. 超高效液相色谱串联质谱法测定腐竹中乌洛托品的含量[J]. 食品工业科技,2014,35(17):298-300.
[3]中华人民共和国卫生部. 食品中可能违法添加的非食用物质和易滥用的食品添加剂名单(第四批)[R]. 2010-3.
[4]国家质量监督检验检疫总局. SN/T 2226-2008 进出口动物源性食品中乌洛托品残留量的检测方法 液相色谱-质谱/质谱法[S]. 北京:中国标准出版社,2008.
[5]黄国春. 气相色谱法测定腐竹中乌洛托品含量的研究[J]. 广西轻工业,2008(6):26-27.
[6]温韬,徐强,张少梅,等. 高效液相色谱法测定腐竹中的乌洛托品[J]. 洛阳理工学院学报:自然科学版,2013,23(3):1-4.
[7]刘春伟,仲雪,马宁. 激光拉曼光谱法快速测定腐竹中的微量乌洛托品[J]. 食品安全质量检测学报,2012,3(4):306-308.
[8]汪辉,夏立新,彭新凯,等. 液相色谱-串联质谱法快速测定食品中的乌洛托品[J]. 食品与机械,2013,29(3):72-78.
[9]周秀云,赵勇,俞婧. SPE-UPLC-MS/MS快速测定豆制品中的乌洛托品[J]. 河北省科学院学报,2013,30(1):64-68.
[10]张爱芝,张书芬,王全林,等.超高压液相色谱-串联质谱法测定腐竹、米线、年糕中的乌洛托品[J].食品安全质量检测学报,2013,4(2):472-478.
[11]陆春良,刘娟,刘向农. 亲水作用液相色谱电喷雾串联质谱法测定米线中乌洛托品残留[J]. 扬州大学学报:农业与生命科学版,2013,34(2):82-86.
[12]Lee SY,Kim BJ,Kim JK. Development of isotope dilution-liquid chromatography tandem mass spectrometry for the accurate determination of fluoroquinolones in animal meat products:Optimization of chromatographic separation for eliminating matrix effects on isotope ratio measurements[J]. Journal of Chromatography A,2013,(1277):35-41.
[13]Hon PYT,Chu PWS,Cheng CH,et al. Development of melamine certified reference material in milk using two different isotope dilution mass spectrometry techniques[J]. Journal of Chromatography A,2011,1218(39):6907-6913.
[14]马雪涛,牛之瑞,冯雷,等. 离子交换固相萃取-超高效液相色谱-串联质谱法测定豆制品中乌洛托品残留量[J]. 食品科学,2014,35(10):166-169.
[15]徐小民,黄百芬,任一平. 豆制品和粉干中乌洛托品的气质联用测定[J]. 食品安全质量检测学报,2013(3):873-876.
Determination of urotropine in bean curd sticks by stable isotopic dilution-liquid chromatography with tandem mass spectrometry
XU Xing1,2,ZHANG Xiao-ming1,*,ZHANG Yan2,SHU Ping2,PENG Fei-jin2
(1.School of Food Science and Technology,Jiangnan University,Wuxi 214122,China;2.Dali Comprehensive Inspection Centre of Quality and Technical Supervision,Dali 671000,China)
A stable isotope dilution mass spectrometry technology for determination of urotropine in bean curd sticks by liquid chromatography with tandem mass spectrometry was developed. Isotopic urotropine was added to bean curd sticks as internal standard,and extracted with acetonitrile. Solid phase extraction(SPE)was applied to purify the sample. The separation was performed on a HILIC column. The analyte was determined by multiple reaction monitoring(MRM)mode with UPLC-MS/MS,and quantified by the internal standard method. The method showed good linear relationship in the range of 1~40 μg/L for urotropine,the correlation coefficient square was 0.9996. The limit of quantitation(LOQ)for sample was 2 μg/kg. The average recoveries were 97.8%~101.5% at spiked levels of 2,10,30 μg/kg. The relative standard deviation(RSD,n=6)was 1.9%~4.8%. This method is sensitive,accurate and reproducible. It is suitable for monitoring illegally added urotropine in bean curd sticks.
stable isotope dilution technology;ultra performance liquid chromatography-tandem mass spectrometry;urotropine;bean curd sticks
2014-11-13
徐幸(1983-),女,在读博士,工程师,研究方向:食品安全质量检测,E-mail:amy911007@163.com。
*通讯作者:张晓鸣(1965-),男,博士,教授,研究方向:食品安全质量控制,E-mail:xmzhang@jiangnan.edu.cn。
云南省卫生厅制修订食品安全地方标准项目(云卫[2014]DB004)。
TS207.3
A
1002-0306(2015)15-0281-04
10.13386/j.issn1002-0306.2015.15.050
