亨廷顿病两家系临床及遗传学分析
2015-05-18孙中武
陈 晶,孙中武
亨廷顿病(huntington disease,HD)又称亨廷顿舞蹈病,是常染色体显性遗传的神经系统退行性病变,临床上以运动障碍、认知损害、精神行为异常三联征为主要特征。该病系因4号染色体短臂4p16.3 IT15基因突变,突变基因产物三核苷酸序列CAG异常重复扩增产生亨廷顿蛋白致病,正常人为11~34个CAG重复序列,CAG异常拷贝超过35个或更多时会发病。CAG重复序列在27~35之间虽不引起疾病,但在减数分裂时容易发生扩增突变,使后代发病;CAG在36~39之间呈年龄相关的不完全外显率,即在随后的时间里,携带者可能很晚发病或不发病;CAG重复序列在40以上,具有完全外显率[1-2]。该研究拟对临床上两个HD家系成员进行IT15基因检测,结合临床症状和认知功能、精神等评价结果,探讨HD的临床特征和CAG重复序列数目对疾病进程的影响。
1 材料与方法
1.1 病例资料 选取安徽医科大学第一附属医院神经内科临床诊断为HD的两个汉族家系,对其家系成员进行详细的临床资料收集、病史调查及IT15基因检测,按照知情和自愿原则,家系1共3代5人参与,家系2中1人参与。4名女性,2名男性,年龄24~80(48.00±20.54)岁。
1.2 研究方法
1.2.1 临床特征调查 详细询问病史,绘制遗传系谱图,尽可能收集受试者的临床资料,包括临床症状和体征、剑桥老年认知量表中文版(cambridge cognitive examination-Chinese version,CAMCOG-C)评分、汉密尔顿抑郁量表评分。两家系遗传系谱图见图1。
1.2.2 IT15基因CAG重复序列测定和CAP计算
抽取两家系6名成员外周静脉血5 ml,采用天根基因组DNA提取试剂盒方法,提取标本中有核DNA,待测DNA的浓度、纯度和体积:DNA浓度应10 ng/μl以上,体积 >100 μl,纯度均应达到:OD260/OD280>1.8,OD260/OD230>1.2;使用 IT15基因特异引物进行扩增;聚合酶链式反应(polymerase chain reaction,PCR)扩增产物进行测序分析。PCR样品使用美国ABI公司遗传分析仪进行测序,测序结果结合人类基因突变数据库HGMD数据进行分析。阳性突变用另一份备用样品重新检测。
根据IT15基因的检测结果,对CAG重复序列>35的受试者计算CAP得分,本文中的CAP公式来源于约翰·霍普金斯大学Ross et al[3]根据几项大型HD数据资源汇集后的标准化CAP得分:CAP=100×AGE×[(CAG-L)÷S],CAG为患者CAG重复序列长度,AGE是患者在检测时的年龄,L和S为常数。S是归一化常数,使CAP得分在患者预测症状出现年龄时接近100。L是一个度量常数,使CAG重复序列长度锚定在相关HD病理分布的低末端。公式中使用的 L和 S数值为30和627,由Warner和Hayden通过再分析Langbehn et al的数据后得到的。
2 结果
2.1 病例资料 家系1先证者,IIa3,女性,80岁。患者在65岁左右开始出现身体不自主晃动,持物稍有不稳,大脚趾不自主扭动,后病情缓慢进展,渐出现性格改变,记忆力下降,身体晃动加重,站立不稳,伴四肢不自主扭动,头部晃动,面部肌肉主要口角四周肌肉不自主抽动,进食困难,无饮水呛咳,近两年体重下降明显。曾按帕金森病予以治疗半年,症状未见缓解反而加重。查体:神清,精神尚可,对答切题,查体部分合作,额纹对称,双瞳孔等大等圆,直径为3 mm,光敏,口周肌肉不自主抽动,站立不稳,颈部、四肢舞蹈样不自主运动,四肢肌张力低,腱反射对称性稍亢,双侧病理征未引出。
IIIa6,男性,55岁,先证者儿子。50左右开始出现走路偶有不稳,手指偶有不自主运动,近两年有性格改变,脾气较前暴躁,自诉不喜社交活动,不爱与人交流,工作没有积极性,年轻时是乐观外向性格。查体:神清,精神可,查体合作,手部偶有不自主运动,四肢肌张力稍增高,肌力正常,腱反射对称,病理征未引出。IIIa8、IIIa10、IVa5本人及家属未发现有运动、精神等方面症状,IIIa10自诉记忆力下降,情绪较前低落。结合基因检测结果和临床症状,根据目前的标准[4],携带CAG异常扩增的IT15等位基因患者发病的定义为:出现无法解释但明确存在的锥体外系行动异常,如舞蹈症、肌张力障碍、运动徐缓及肌强直。IIa3、IIIa6已发病,处于症状期,IIIa8、IIIa10处于临床诊断前期,IVa5为正常人。
家系2中先证者IVb2,女性,24岁。半年前因在饭店做上餐员经常打碎盘子,无法继续工作失业在家,家人发现性格改变,不喜出门及与人交流、自闭,担心有抑郁症就诊于精神科,后精神科医师建议神经科就诊,接诊时查体:神清,精神可,对答切题,查体合作,额纹对称,面部肌肉偶有不自主抽动,四肢肌张力稍减低,腱反射亢进,指鼻试验阳性,闭目难立征阴性,双侧病理征未引出。结合基因检测结果和临床症状,IVb2尚未达到临床诊断发病标准,为临床诊断前期。先证者母亲IIIb3,据家属回忆在25岁左右发病,起初因动作迟缓,精细动作笨拙被家人发现就医,一直未予明确诊断,后患者病情进行性加重,出现四肢及脸部不自主运动,后渐渐四肢强直,无法行走,说话困难,吞咽困难,45岁病逝。
2.2 基因检测结果 按照自愿原则抽取两家系6人外周血进行IT15基因检测,基因检测结果见图2。其中家系1先证者IIa3(CAG)n拷贝数为17/42,其子女为IIIa6、IIIa8、IIIa10(CAG)n拷贝数分别为18/42、18/42、20/42,其孙 IVa5(CAG)n 拷贝数为18/19。家系2先证者IVb2(CAG)n拷贝数为20/53。见表1。
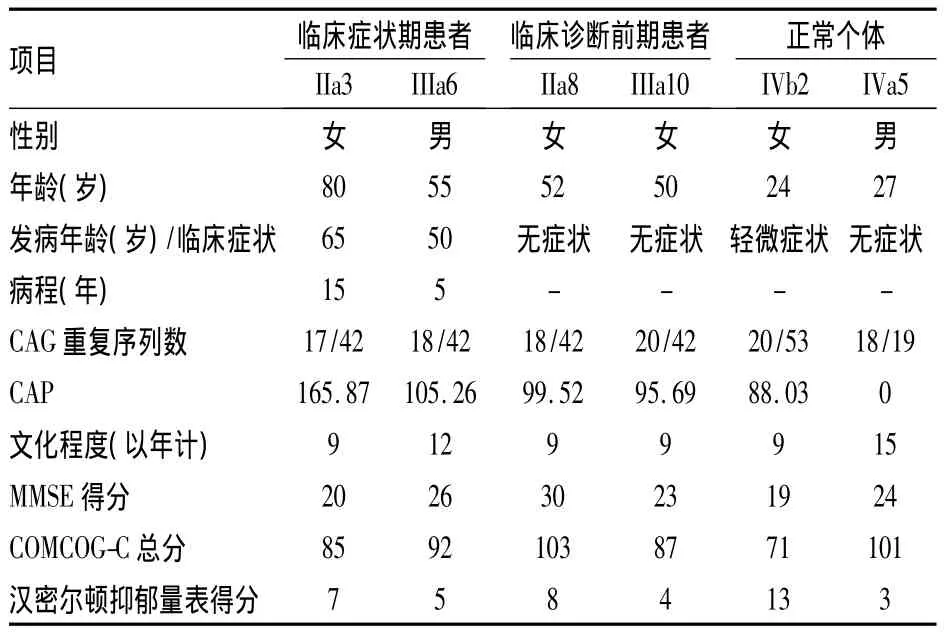
表1 两HD家系受试者临床资料
3 讨论
1个突变HD基因携带者在临床诊断发病前,没有任何症状,可以像正常人一样生活,然后会经历一个明显症状前期,以轻微的运动、认知、行为、个性改变为特征[5]。一旦开始发病,患者的运动、认知、行为等症状将稳固下降[6],临床诊断和发病年龄根据临床典型运动症状的出现决定。
本研究家系2中IVb2年仅24岁,CAG重序列数是53,较其他临床诊断前期患者有明显认知损害和人格改变,与文献[7]报道的CAG重复序列越多,发病年龄越早且临床症状越重一致。IIIa8、IIIa10、IVb2虽同在临床诊断前期,但3人表现也各不同。IIIa8未发现任何运动、精神症状,COMCOG-C认知评估和汉密尔顿抑郁量表评估也正常,IIIa10有轻微认知损害和情绪改变,IVb2运动症状不明显,CAMCOG-C评分71分,有明显认知障碍,汉密尔顿抑郁量表13分,提示有抑郁可能,且曾有自杀倾向,人格改变明显,这和现有研究[8]认为患者的认知损害在运动症状出现前的数十年里就存在相符。大型的临床研究Predict-HD study[9]数据显示,在典型运动症状出现前的15~20年间,HD基因携带者可出现相关改变,如纹状体萎缩、认知的改变、行为异常、抑郁等。一个基于多中心2 318个样本COHORT研究也显示,亨廷顿相关改变在临床诊断之前就开始出现,和对照组相比,HD突变基因携带者在UHDRS关于认知部分的评分和MMSE评分更低,体重下降更明显,有更多的自杀倾向[10]。
CAG重复序列数和年龄是两个描绘疾病进展特征的重要变量,大量的研究[11]表明,HD的发病年龄与CAG重复序列数有关。近年来很多学者在寻找疾病进展的标志物,以用来预测HD的发病年龄和用于以后HD疾病治疗的大规模临床研究。1997年,Penney et al[12]通过对 HD 患者进行尸检,发现了HD患者在死亡时纹状体的病理改变和CAG重复序列有关,提出纹状体功能障碍=AGE×(CAG-L),其中AGE是患者目前的年龄、CAG是基因重复序列数、L为常数,该公式反映了基因产物的毒性积累。后来又有学者更倾向于CAG的年龄产物CAP的概念[13]。本文中使用的 CAP 计算公式[1]来自于几项大型HD数据资源汇集后的标准化CAP得分,使CAP得分在患者预测症状出现年龄时接近100。本研究中 IIa3、IIIa6 CAP得分大于100,与临床已发病符合,IIIa8、IIIa10、IVb2 CAP得分小于100,提示在临床诊断前期,与临床结果符合。亨廷顿舞蹈病的临床表现和疾病进展与CAG重复序列和年龄有关,CAP作为综合了这两个变量的指标,具有重要的临床意义,不仅可以用于评估患者发病年龄,更重要的是在大型临床研究中,根据CAP得分,按照不同的预测发病年龄可将人群进行分层,有助于研究者更好的了解亨廷顿病的疾病进程,为日后针对疾病治疗和干预的研究提供帮助。
HD尚无有效的方法阻止疾病进展,目前仍以对症治疗为主[14]。家系2中的先证者,现已80岁,病程十余年,不自主运动明显,给以氟哌啶醇治疗后症状明显得到控制,生活质量提高。家系2中的先证者,现24岁,刚开始出现症状,临床症状轻微,未予任何药物治疗,经过家庭护理和心理疏导后,情绪改善明显。由于舞蹈病有着不同的临床表现,治疗上要求综合多方面的,包括神经科医师、精神科医师、护士、社会工作者、遗传工作者、理疗师、语言治疗师、营养师。而且随着疾病的进展,在不同的阶段患者表现不同,这就要求治疗方案随着病程进展而灵活改变,治疗是一个综合长期的过程[15]。
[1]Squitieri F,Jankovic J.Huntington’s disease:how intermediate are intermediate repeat lengths?[J].Mov Disord,2012,27(14):1714-7.
[2]Killoran A,Biglan K M,Jankovic J,et al.Characterization of the Huntington intermediate CAG repeat expansion phenotype in PHAROS[J].Neurology,2013,22(80):2022 - 7.
[3]Ross C A,Aylward E H,Wild E J,et al.Huntington disease:natural history,biomarkers and prospects for therapeutics[J].Nat Rev Neurol,2014,10(4):204 -16.
[4]Sturrock A,Leavitt B R.The clinical and genetic features of huntington disease[J].J Geriatric Psychiatry Neurol,2010,23(4),243-59.
[5]Tabrizi S J,Langbehn D R,Leavitt B R,et al.Biological and clinical manifestations of Huntington’s disease in the longitudinal TRACK-HD study:cross-sectional analysis of baseline data[J].Lancet Neurol,2009,8(9):791 -801.
[6]Dorsey E R,Beck C A,Darwin K,et al.Natural history of huntington disease[J].JAMA Neurol,2013,70(12):1520 -30.
[7]Shoulson I,Young A B.Milestones in huntington disease[J].Mov Disord,2011,26(6):1127 -33.
[8]Paulsen J S.Cognitive impairment in huntington disease:diagnosis and treatment[J].Curr Neurol Neurol Rep,2011,11(5):474-83.
[9]Paulsen J S,Long J D,Johnson H J,et al.Clinical and biomarker changes in premanifest huntington disease show trial feasibility:A decade of the PREDICT-HD study[J].Front Aging Neurosci,2014,22(6):78.
[10]Huntington study group COHORT investigators,Dorsey E.Characterization of a large group of individuals with huntington disease and their relatives enrolled in the COHORT study[J].PLoS One,2012,7(2):e29522.
[11]Langbehn D R,Hayden M R,Paulsen J S,et al.CAG-repeat length and the age of onset in huntington disease(HD):A review and validation study of statistical approaches[J].Am J Med Genet B Neuropsychiatr Genet,2010,153B(2):397 -408.
[12]Penney J B Jr,Vonsattel J P,MacDonald M E,et al.CAG repeat number governs the development rate of pathology in Huntington's disease[J].Ann Neurol,1997,41(5):689 - 92.
[13]Zhang Y,Long J D,Mills J A,et al.Indexing disease progression at study entry with individuals at-risk for Huntington disease[J].Am J Med Genet B Neuropsychiatr Genet,2011,156B(7):751 -63.
[14]Cummins A,Eggert J,Pruitt R,et al.Huntington disease:Implications for practice[J].Nurse Pract,2011,36(2):41.
[15]Singer C.Comprehensive treatment of huntington disease and other choreic disorders[J].Cleve Clin J Med,2012,79(2):S30 -4.
