戈谢病2 例
2015-04-28张晓胜史敏
张晓胜 史敏
戈谢病(gaucher disease)是一种葡萄糖脑苷脂酶(GBA)缺乏引起的常染色体隐性遗传性疾病[1]。临床表现为多系统异常包括外周血细胞减少、肝脾肿大[2]以及骨质损害、神经系统异常。此病可发生于从幼儿到老年的任何年龄,症状从轻度血小板减少症到脾大或缺血性坏死[3]。最常见的表现是症状性脾肿大或血小板减少症,因此患者常以血液系统疾病就诊。本文介绍两例无明显临床症状的患者,由于出现不同感染去医院就诊,经检查发现血小板减少和脾肿大,进一步骨穿发现戈谢细胞而确诊为GD。因此加强GD细胞形态学认识对防止GD 漏诊和误诊有非常重要的作用。
1 病例资料
例1,女,27 岁,因腰痛、尿频就诊于当地医院,当时无发热、无尿中泡沫增多,无夜尿增多及肉眼血尿,无尿急、尿痛,当地考虑为泌尿系感染,同时查体发现脾脏增大,当地医院给予抗感染治疗后腰痛、尿频症状好转,为进一步明确脾脏肿大原因就诊于我院。门诊行骨穿,结果回报后考虑戈谢病,无智力障碍,无胸骨压痛、无腹痛、腹泻,无出血倾向。大便正常、无血便、黑便等,体重无明显减轻。体检:发育正常,体型偏瘦,神志清楚,T 36.9 ℃,P 80 次/min,R 20 次/min,Bp 121/74 mm Hg。全身皮肤无黄染、出血点及瘀斑,浅表淋巴结未及肿大。头颅无畸形,眼睑无浮肿,结膜无苍白,巩膜无黄染,两侧瞳孔正大等圆,对光反射灵敏,耳鼻无畸形及异常分泌物。双肺呼吸音清,未闻及干、湿性啰音及胸膜摩擦音,心律整,未闻及杂音。腹平坦,无压痛、反跳痛及肌紧张,未触及包块。肝肋下可触及边缘,脾肋下10 cm,质中偏硬,无触痛;肝区、双肾区无叩击痛,肠鸣音正常存在。肛门及外生殖器未见异常。脊柱四肢无畸形,活动自如。
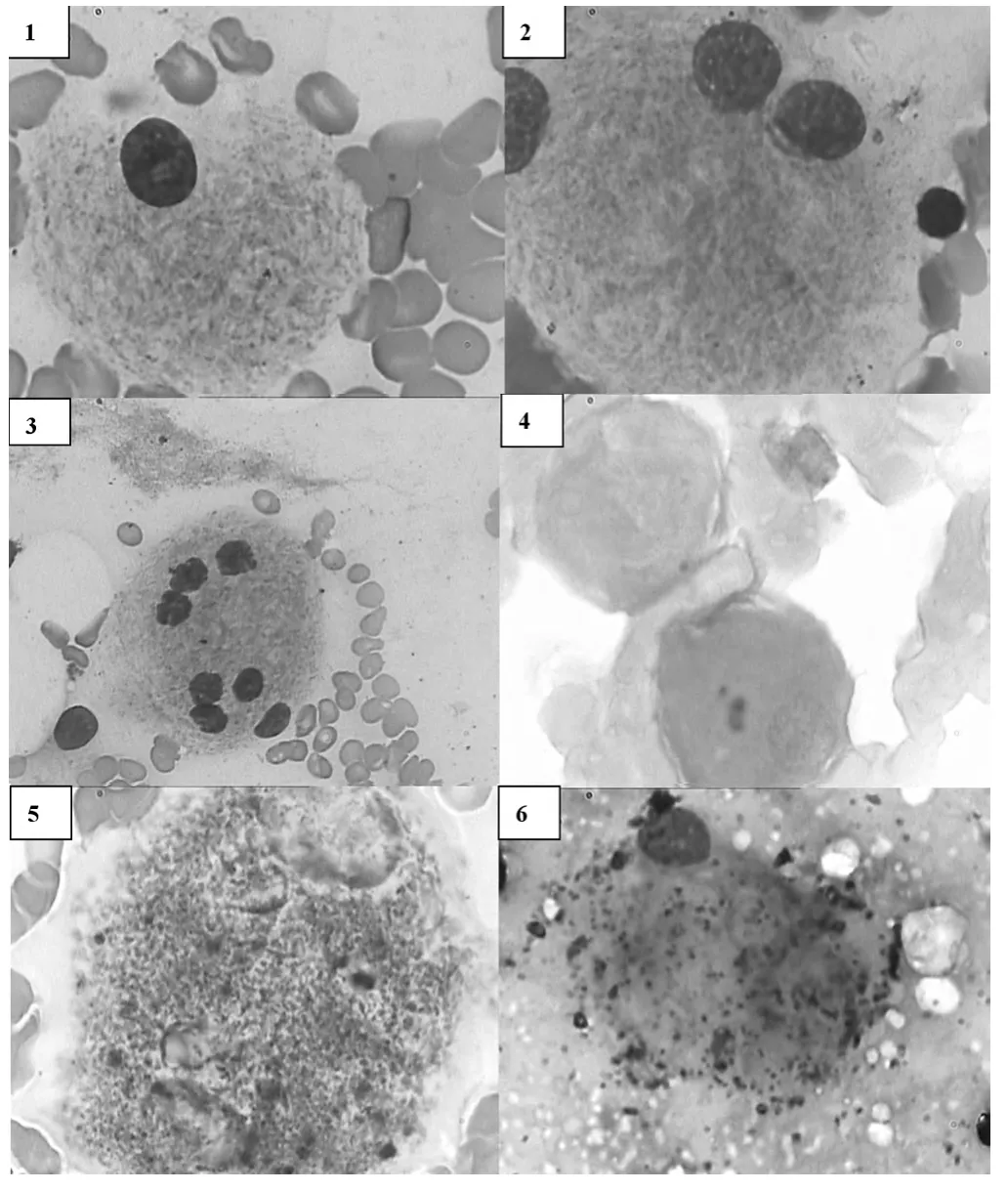
血常规:WBC 2.4 × 109/L,Hb 106 g/L,PLT 51 g/L。骨髓细胞形态学检查:有核细胞增生活跃,片尾易见戈谢细胞,占3%,该细胞体积较大,形态不规则,胞核小,偏于一侧,呈圆形、椭圆形或不规则形,可见双核、多核,染色质粗糙,偶见核仁,胞质量丰富,染蓝色或灰红色,呈波纹状或洋葱皮样结构(图1 ~3)。细胞化学染色:PAS(+)(图4),POX(-),ACP(+)(图5),酒石酸抑制(-),SBB 弱(+)(图6),NAP(- ),铁染色(+ )(图 7)。β-葡萄糖苷酶2.0 nmol·h-1·mg-1Pr(正常范围:6.0 ~16.7),β-半乳糖苷酶149.7 nmol·h-1·mg-1Pr(正常范围:88 ~220)。肝炎筛查:HBS Ab (+ ),铁蛋白400.5 ng/ml,免疫球蛋白IgG 20.5 g/L,IgM 3.70 g/L。腹部B 超:肝大,巨脾、脾门静脉增宽,余未见异常。上消化道造影:胃炎。临床诊断戈谢病。家属自动要求前往上级医院诊治。

例2,女,5 岁,主因发现腹部肿块1 月余、流涕、轻咳2 d入院。查体T 36.5℃,P 92 次/min,R 22 次/min,无贫血貌,无皮肤出血点及瘀斑,浅表淋巴结不大,咽充血,扁桃体不大。心肺查体未见异常,腹部膨隆,无压痛,脾肋下约7.5 cm 可及,质硬,肝肋下未及,肠鸣音正常存在。双下肢无水肿,活动自如。神经系统查体未见阳性体征。
血常规:WBC 2.52 × 109/L,Hb 112 g/L,PLT 62 g/L。骨髓细胞形态学检查:有核细胞增生明显活跃,涂片中多见戈谢细胞。细胞化学染色:POX 染色阴性,SBB 阴性,PAS 强阳性,ACP 强阳性。生化检查:ALT 15 U/L,AST 43 U/L,ALP 134 U/L。肾功能正常。β-葡萄糖苷酶3.1 nmol·h-1·mg-1Pr(正常范围:6.0 ~16.7),β-半乳糖苷酶109.3 nmol·h-1·mg-1Pr(正常范围:88 ~220)。腹部B 超:巨脾、脾门静脉增宽,腹腔多发淋巴结肿大,肝、胆、胰、双肾未见占位性病变。临床诊断戈谢病。给予抗感染对症支持治疗,家属要求前往上级医院诊治。
2 讨论
戈谢病为常染色体隐性遗传病,是由于β-葡萄糖脑苷脂酶缺乏导致葡萄糖脑苷脂在肝、脾、骨髓等处沉积的一种少见的遗传性糖脂代谢异常疾病。Brady 等在1964 年证明溶酶体酸性β-葡萄糖脑苷脂酶(gluccocerebrosidase,GC 酶)缺乏引起葡萄糖脑苷脂的贮积。该病由于基因突变导致机体GC 苷酶活性缺乏,形成贮积细胞即戈谢细胞,导致受累组织器官出现病变[2]。临床上以肝脾肿大,全血细胞减少,智力低下,反复癫痫和共济失调、骨骼受累为主要表现。根据戈谢病发病的急缓和内脏受累程度及有无神经系统将其分为三种类型:慢性型(Ⅰ型、非神经型、成人型)、急性型(Ⅱ型,神经型,婴儿型)和亚急性型(Ⅲ型,神经型,幼儿型)。
本病在我国发病率较低,北方多于南方[4]。近年来国内3 型均有报道,虽以儿童和青少年为主[5,6],但仍以Ⅰ型多见。Ⅰ型为最常见的类型,儿童和成人均可发病,无神经系统受累症状,预后较好。起病缓慢,病程长,轻型可无症状,在任何年龄均可发现脾大。重型在儿童期发病,进展缓慢,常以肝脾大和贫血就诊。骨骼受损以股骨下端受累最为明显。Ⅱ型及Ⅲ型患者除有与I 型相似的肝脾肿大、贫血、血小板减少等表现外,均有神经系统受累,如共济失调、精神错乱、行为改变、脑电图广泛异常,亦常见骨骼病变及病理性骨折。2 例患者均脾大,白细胞、血小板减低,但不贫血,症状较轻,骨髓中出现戈谢细胞,符合Ⅰ型特点。此病以对症治疗为基础,有条件者可进行酶替代治疗。
由于戈谢病多以脾肿大及全血细胞减少为主要表现,可能会使临床误诊为脾功能亢进。故对原因不明的脾肿大,外周血三项减少者应及时作全面、详细的检查。本病诊断主要依靠患者白细胞或成纤维细胞中葡萄糖脑苷脂酶活性明显缺乏和骨髓或脾穿刺涂片找到戈谢细胞确定,只要熟悉戈谢细胞的形态学特征,诊断本病并不困难。组化染色中戈谢细胞的PAS(+ + ++)、ACP(+ +)、SB(-)、POX(-)、ALP(-),可以将戈谢细胞与类似细胞如尼曼-匹克细胞、海蓝细胞相鉴别,尼曼-匹克细胞ACP 阴性,PAS 阴性;海蓝细胞ACP 阴性,PAS 强阳性。本例患者PAS 和ACP 均阳性,符合戈谢细胞特征。高血脂症、急慢性粒细胞白血病、泛发的神经节苷脂沉着及多发性骨髓瘤可出现类戈谢细胞,需要通过临床表现加以鉴别。
GBA 基因突变类型已报道近300 种。不同种族其常见的突变等位基因的比例不同,其临床表现、病程及治疗效果也不同。到目前为止已发现中国人GD 突变类型40 种,L444P 为最常见的突变类型,其次为F213I、N188S、V375L 和M416V 突变类型,与亚洲地区的报道相似[7],而与高加索人、犹太人不同,如超过70%的Ashkenazi Jewish 人突变类型为N370S[8]。
1 Brady RO,Kanfer JN,Shapiro D.Metabolism of glucocerebrosides.II.Evidence of an enzymatic deciency in Gaucher’s disease.Biochemical and Biophysical Research Communications,1965,18:221-225.
2 Cox TM,Scho eld JP.Gaucher’s disease:clinical features and natural history.Baillieres Clinical Haematology,1997,10:657-689.
3 Thomas AS,Mehta A,Hughes DA.Gaucher disease:haematological presentations and complications.British Journal of Haematology,2014,165:427-440.
4 丛玉隆,李顺义,卢兴国主编.中国血细胞诊断学.第1 版.北京:人民军医出版社,2010.310-314.
5 王秀敏,梁黎.戈谢病12 例临床分析.浙江预防医学,2005,17:43-50.
6 王珏,顾中华,李天宇.幼年型戈谢病一例.中华检验医学杂志,2009,32:493.
7 Yoshikatsu E,Hiroyuki I.Clinical and molecular characteristics of Japanese Gaucher Disease.Neurochemical Research,1999,24:207-211.
8 Beutler E,Nguyen NJ,Henneberger MW,et al.Gaucher disease:gene frequencies in the Ashkenazi Jewish population.Am J Hum Genet,1993,52:85-88.
