11-deoxyheloside A的合成*
2015-03-20张雪微张秀丽
张雪微, 王 鹏, 宋 妮, 张秀丽, 王 聪, 李 明
(中国海洋大学医药学院海洋药物教育部重点实验室,山东 青岛 266003)
11-deoxyheloside A的合成*
张雪微, 王 鹏, 宋 妮, 张秀丽, 王 聪, 李 明**
(中国海洋大学医药学院海洋药物教育部重点实验室,山东 青岛 266003)
首次完成天然胆甾烷型皂苷11-deoxyheloside A的合成。在相转移催化的条件下,葡萄糖溴苷1与膦酸单乙酯2反应以69%的收率得到葡萄糖膦酸酯供体3;以胆甾烷苷元4为原料,经Luche还原和乙酰化反应以及三氟甲磺酸负载硅胶脱除TBDPS基团得到胆甾烷二醇10;在金催化剂的作用下,胆甾二醇10与葡萄糖膦酸酯供体3进行糖苷化反应,然后脱除酰基保护基以97%的产率完成11-deoxyheloside A的合成。
11-deoxyheloside A;葡萄糖膦酸酯;金催化糖苷化
皂苷是一类具有多种药效活性的糖缀合物,广泛存在于传统药用植物中,具有抗肿瘤、抗病毒、抗菌、抗炎、抗寄生虫、免疫调节等多种生物活性[1]。黄地百合(Chamaeliriumluteum(L.) A. Gray)的根和茎是传统的中药材,用于治疗女性生殖系统疾病[2]。至2012年,已从黄地百合中分离得到一系列开链甾体皂苷[2](见图1),但由于它们的含量低微以及分离提取困难,限制了其构效关系和深入的药理活性研究。11-deoxyheloside A(见图1)作为黄地百合胆甾烷皂苷的代表,至今未见对其的化学合成报道。本文以易得的胆甾烷苷元4和葡萄糖溴苷1为原料,采用金催化的邻炔基膦酸酯糖苷化方法完成了11-deoxyheloside A的全合成,为合成黄地百合中系列胆甾烷型皂苷奠定了基础。
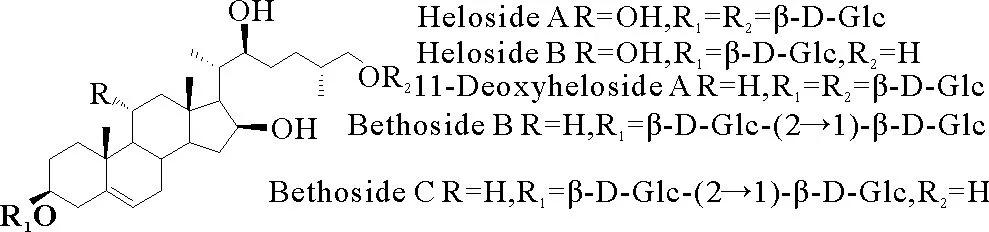
图1 11-deoxyheloside A和相关皂苷的结构
1 实验部分
1.1 仪器与试剂
试剂 TBDPSCl(叔丁基二苯基氯硅烷),NaBH4,DIPEA(二异丙基乙基胺)等均为阿拉丁试剂公司产品,直接用于反应。胆甾烷苷元4为实验室已有原料。未经特殊说明,其余所用的试剂为国产分析纯。1,2-二氯乙烷、CH2Cl2经过CaH2回流重蒸处理,所用石油醚为60~90℃沸程。硅胶柱层析所用硅胶购自青岛海洋化工厂分厂,规格为300~400目。
仪器 JEOLJNM-ECP 600 MHz和Bruker ARX500 (500 MHz)核磁共振波谱仪; JASCO P-1020自动比旋光仪; 85-2型恒温磁力搅拌器; 旋转蒸发仪(BUCHI, R-114:EYELA, DTC- 21)。
1.2 实验操作
1.2.1 化合物3的合成 膦酸单乙酯2 (0.162 g, 0.61 mmol)溶于CHCl3(40 mL)中,加入K2CO3(0.169 g, 1.22 mmol), 甲基三辛基氯化铵(0.296 g, 0.732 mmol)和H2O (40 mL), 40 ℃下剧烈搅拌,分3次加入葡萄糖溴苷1(0.6 g, 0.93 mmol),TLC检测反应完全。分出有机相,加入CHCl3稀释。依次用饱和NaHCO3水溶液, 5% Na2S2O3水溶液和饱和食盐水洗涤,无水Na2SO4干燥。抽滤、浓缩后硅胶柱层析纯化(Petroleum ether:EtOAc=4:1-2:1)得到淡黄色泡沫状固体3 (356 mg, 0.42 mmol, 69%); Rf0.10 (Petroleum ether:EtOAc=2:1):1H NMR (600 MHz, CDCl3)δ8.07 (dd,J=8.4,1.3 Hz, 2H), 8.01-7.99 (m, 3H), 7.95-7.87 (m, 11H), 7.84-7.79 (m, 6H), 7.68-7.65 (m, 2H), 7.58-7.25 (m, 41H), 7.22 (dd,J=8.3, 7.5 Hz, 2H), 7.11-7.04 (m, 4H), 6.92-6.88 (m, 1H), 5.92-5.82 (m, 6H),5.78-5.63 (m, 6H), 4.70 (dd,J=12.2, 2.9 Hz, 1H), 4.52 (dd,J=12.2, 5.1 Hz, 1H), 4.41 (dd,J=12.1, 3.0 Hz, 2H), 4.37-4.33 (m, 1H), 4.30 (dd,J=12.1, 5.2 Hz, 2H), 4.20-4.16 (m, 4H), 3.97-3.90 (m, 2H), 3.88-3.80 (m, 2H),2.49-2.43 (m, 6H), 1.64-1.59 (m, 6H), 1.52-1.46 (m, 6H), 1.27 (t,J=7.1 Hz, 3H), 1.01-0.93 (m, 15H);13C-NMR (150 MHz, CDCl3)δ165.9, 165.8, 165.6, 165.1, 165.0, 164.9, 164.7, 133.45, 133.40, 133.36, 133.27, 133.22, 133.18, 133.12, 133.0, 132.9, 132.77, 132.72, 131.9, 131.8, 129.81, 129.78, 129.72, 129.71, 129.6, 129.48, 129.43, 128.9, 128.7, 128.6, 128.5, 128.38, 128.36, 128.24, 128.22, 128.0, 126.9, 126.8, 126.7, 126.64,126.60,126.4,126.3,96.9,96.8,95.7 (d,J=4.7 Hz), 95.2 (d,J=5.8 Hz), 78.6 (d,J=6.3 Hz), 78.4 (d,J=6.7 Hz), 72.9, 72.8, 72.70, 72.68, 71.8 (d,J=7.9 Hz), 71.5 (d,J=8.4 Hz), 69.2, 69.1, 62.64, 62.56, 62.5 (d,J=1.7 Hz), 62.3 (d,J=7.0 Hz), 30.5, 30.4, 22.0, 21,9, 19.4, 19.3, 16.0, 15.9 (d,J=7.2 Hz), 15.8 (d,J=6.7 Hz), 13.7, 13.6;31P NMR (243 MHz, CDCl3)δ18.29, 17.90; ESI-HRMS calcd for C48H46O12P[M+H]+845.2721, Found 845.2705。
1.2.2 化合物5的合成 化合物4 (0.6 g, 0.79 mmol)溶于MeOH和CH2Cl2(11 mL,v:v=9:2)的混合溶剂中,加入MeONa (8.6 mg, 0.16 mmol),室温反应过夜,TLC显示反应完全。Dowex-50(H+)树脂调pH至中性。过滤、浓缩,硅胶柱层析纯化(Petroleum ether:AcOEt=2.5:1)得到白色泡沫状固体5 (0.49 g, 0.69 mmol, 87%):1H NMR (500 MHz, CDCl3)δ7.70-7.64 (m, 4H), 7.43-7.33 (m, 6H), 5.10 (d,J=5.2 Hz, 1H), 4.95 (td,J=8.0, 4.9 Hz, 1H), 3.56-3.48 (m, 1H), 3.41 (d,J=5.8 Hz, 1H), 2.97-2.91 (m, 1H), 2.65-2.58 (m, 1H), 2.43-2.26 (m, 1H), 2.15-2.09 (m, 1H), 1.95 (s, 3H), 1.92-1.80 (m, 4H), 1.76-1.64 (m, 5H), 1.63-1.15 (m, 9H), 1.12 (d,J=7.1 Hz, 3H), 1.05 (s, 9H), 0.98 (s, 3H), 0.90 (d,J=6.8 Hz, 3H), 0.84 (s, 3H).
1.2.3 化合物6的合成 化合物5 (0.2 g, 0.28 mmol),咪唑(0.038 g, 0.56 mmol)溶于DMF(4 mL)中,加入TBDPSCl (0.091 mL,0.365 mmol),室温反应6 h,TLC检测反应完全。蒸出DMF,加入饱和NH4Cl水溶液洗涤,CH2Cl2萃取,合并有机相。依次用饱和NaHCO3水溶液、蒸馏水、饱和食盐水洗涤,无水Na2SO4干燥。抽滤、浓缩后硅胶柱柱层析(Petroleum ether: CH2Cl2=1:1-1:1.5→Petroleum ether:AcOEt =20:1)得白色固体6 (0.232 g, 0.25 mmol, 90%):[α]25D-0.85(c1.2, CHCl3);1H NMR (600 MHz, CDCl3)δ7.69-7.63 (m, 8H), 7.43-7.34 (m, 12H), 5.10 (d,J=5.2 Hz, 1H), 4.95- 4.91 (m, 1H), 3.54-3.48 (m, 2H), 3.44 (dd,J=9.9, 6.1 Hz, 1H), 2.92-2.87 (m, 1H), 2.59-2.52 (m, 1H), 2.39-2.25 (m, 3H), 2.13-2.10 (m, 1H), 1.90 (m, 3H), 1.88-1.82 (m, 3H), 1.71-1.57 (m, 5H), 1.50-1.38 (m, 4H), 1.35-1.26 (m, 1H), 1.22-1,17 (m, 1H), 1.08 (d,J=6.0 Hz, 3H), 1.06 (s, 9H), 1.05 (s, 9H), 0.99 (s, 3H), 0.90 (d,J=5.6 Hz, 3H), 0.83 (m, 3H);13C NMR (125 MHz, CDCl3)δ213.2, 169.7, 141.2, 135.7, 135.6, 134.8, 134.7, 133.9, 129.5, 129.46, 129.42, 127.6, 127.5, 127.4, 120.80, 75.7, 73.1, 68.7, 55.0, 54.0, 49.8, 43.5, 42.4, 41.8, 39.7, 38.9, 37.1, 36.4, 35.5, 34.8, 31.8, 31.6, 31.2, 27.0, 26.9, 21.1, 20.7, 19.4, 19.3, 19.1, 16.8, 16.7, 13.2; ESI-HRMS calcd for C61H82O5NaSi2[M+Na]+973.5593, Found 973.5576。
1.2.4 化合物7和8的合成 化合物6 (0.3 g, 0.32 mmol)溶于THF(40 mL)中,冰浴下,加入NaBH4(1.8 g, 47 mmol)和CeCl3·7 H2O (3.5 g, 9.6 mmol),缓慢升至室温反应过夜,TLC检测反应完全。CH2Cl2提取有机物,浓缩,硅胶柱层析(Petroleum ether:AcOEt=9:1)得到7(22 mg, 0.024 mmol, 18%)和8 (88 mg, 0.096 mmol, 74%);7:[α]25D-4.7(c0.73, CHCl3);1H NMR (500 MHz, CDCl3)δ5.19 (m, 1H), 5.10 (d,J=5.2 Hz, 1H), 3.56-3.48 (m, 2H), 3.44 (dd,J=9.9, 6.1 Hz, 1H), 3.40 (t,J=6.6 Hz, 1H), 2.41-2.26 (m, 2H), 2.14-2.12 (m, 1H), 1.91 (s, 3H), 1.06 (s, 9H), 1.04 (s, 9H), 0.99 (s, 3H), 0.91 (d,J=6.8 Hz, 1H), 0.89 (d,J=7.1 Hz, 1H), 0.86 (s, 3H);13C NMR (125 MHz, CDCl3)δ170.4, 143.1, 141.3, 135.8, 135.7, 135.60, 135,58, 134.74, 133.9, 130.9, 129.5, 129.44, 129.43, 129.41, 128.8, 127.6, 127.44, 127.42, 120.8, 75.3, 73.2, 72.9, 68.6, 65.6, 55.6, 54.6, 49.9, 42.4, 42.3, 39.6, 37.1, 36.4, 35.8, 35.4, 35.1, 34.0, 31.8, 31.6, 31.4, 29.7, 29.4, 27.0, 26.9, 21.3, 20.7, 19.4, 19.3, 19.1, 16.9, 12.9, 12.5; 8:[α]25D-1.03 (c1, CHCl3);1H NMR (600 MHz, CDCl3)δ7.69-7.63 (m, 8H), 7.43-7.34 (m, 12H), 5.10 (d,J=2.7 Hz, 1H), 5.03-4.98(m, 1H), 3.57-3.40 (m, 4H), 2.36-2.29 (m, 2H), 2.16-2.11 (m, 2H), 2.02 (s, 3H), 1.06 (s, 9H), 1.04 (s, 9H), 0.99 (s, 3H), 0.94 (d,J=5.8 Hz, 3H), 0.91 (d,J=5.6 Hz, 3H), 0.87 (s, 3H);13C NMR (125 MHz, CDCl3)δ170.6, 141.3, 135.77, 135.76, 135.61, 135.59, 134.80, 134.75, 134.0, 129.5, 129.46, 129.42, 127.6, 127.5, 127.4, 120.8, 74.9, 73.1, 69.0, 56.1, 54.5, 49.9, 43.4, 42.8, 42.4, 39.6, 37.1, 36.7, 36.5, 35.8, 35.2, 31.8, 31.6, 31.4, 30.6, 28.0, 27.0, 26.9, 21.3, 20.7, 19.4, 19.3, 19.1, 17.0, 12.5, 12.1; ESI-HRMS calcd for C61H84O5NaSi2[M+Na]+975.5749, Found 975.5745。
1.2.5 化合物9的合成 化合物 7 (23 mg, 0.024 mmol) 溶于吡啶(1 mL)中,加入Ac2O (0.071 mL, 0.75 mmol),室温反应过夜,TLC显示反应完全。蒸出吡啶,CH2Cl2稀释,依次用1M HCl、蒸馏水、饱和食盐水洗涤,无水Na2SO4干燥。抽滤、浓缩,硅胶柱层析(Petroleum ether :AcOEt=20:1)得化合物9 (22 mg, 0.022 mmol, 92%):[α]25D-1.15 (c0.8, CHCl3);1H NMR (500 MHz, CDCl3)δ7.69-7.62 (m, 8H), 7.43-7.34 (m, 12H), 5.18- 5.15 (m, 1H), 5.10 (d,J=5.1 Hz, 1H), 4.81 (t,J=6.5 Hz, 1H), 3.56-3.50 (m, 1H), 3.46-3.42 (m, 2H), 2.45-2.35 (m, 1H), 2.32 (t,J=11.8 Hz, 1H), 2.14-2.08 (m, 2H), 2.00 (s, 3H), 1.93 (s, 3H), 1.06 (s, 9H), 1.04 (s, 9H), 0.98 (s, 3H), 0.97 (d,J=8.6 Hz, 3H), 0.89 (d,J=6.7 Hz, 3H), 0.84 (s, 3H);13C NMR (125 MHz, CDCl3)δ170.8, 170.0, 141.2, 135.7, 135.6, 134.8, 134.7, 133.90, 133.87, 129.5, 129.45, 129.40, 127.6, 127.45, 127.41, 120.84, 75.8, 75.6, 73.1, 68.6, 55.9, 54.4, 49.9, 42.4, 42.3, 39.6, 37.1, 36.4, 35.7, 35.2, 33.2, 31.8, 31.5, 31.4, 29.9, 29.7, 29.4, 27.0, 26.8, 21.3, 21.1, 20.7, 19.4, 19.3, 19.1, 16.7, 12.4, 12.2; ESI-HRMS calcd for C63H86O6NaSi2[M + Na]+1017.5855, Found 1017.5838。
化合物8 (178 mg, 0.19 mmol)溶于CH2Cl2(6 mL)中,加入DMAP (0.046 g, 0.37 mmol)、DIPEA (0.099 mL, 0.57 mmol)和4Å MS (600 mg),室温下搅拌1 h, 移入-10 ℃下继续搅拌30 min,加入MsCl (0.029 mL, 0.37 mmol),继续反应,TLC检测反应完全。过滤掉分子筛,AcOEt稀释,加入冰水,分出有机相。AcOEt萃取,合并有机相,依次用蒸馏水、饱和食盐水洗涤,无水Na2SO4干燥。抽滤、浓缩得黄色固体,不经纯化直接投入下步反应。取DBU (0.057 mL, 0.38 mmol)溶于甲苯(6 mL)中,加入AcOH(0.043 mL, 0.76 mmol),室温搅拌反应30min后,加入制备甲磺酸酯,95 ℃反应11 h,TLC检测反应完全。反应液冷至室温,依次用2M HCl溶液、10%的K2CO3水溶液、蒸馏水、饱和食盐水洗涤,无水Na2SO4干燥。抽滤、浓缩,硅胶柱层析(Petroleum ether:AcOEt=65:1-20:1)得到化合物9 (74 mg, 0.072 mmol,39%)。
1.2.6 化合物10的合成 化合物9 (32 mg, 0.032 mmol)溶于MeCN和CH2Cl2(0.5 mL, v:v=3:2)中,加入TfOH-SiO2(12 mg, 0.024 mmol),50 ℃搅拌反应1 h,反应完全。过滤掉硅胶,CH2Cl2淋洗硅胶,浓缩,硅胶柱层析(Petroleum ether:AcOEt=1.5:1)得到化合物10 (15 mg, 0.029 mmol, 91%):[α]25D+4.1 (c1, CHCl3);1H-NMR (500 MHz, CDCl3)δ5.33 (d,J=4.9 Hz, 1H), 5.24-5.20 (m, 1H), 4.83 (t,J=6.9 Hz, 1H), 3.53-3.51 (m, 1H), 3.48-3.39 (m, 2H), 2.46-2.09 (m, 6H), 2.04 (s, 3H), 2.02 (s, 3H), 1.00 (s, 3H), 0.99 (d,J=6.7 Hz, 3H), 0.91 (d,J=6.7 Hz, 3H), 0.87 (s, 3H);13C NMR (125 MHz, CDCl3)δ170.9, 170.1, 140.7, 121.4, 75.8, 75.7, 75.6, 71.7, 56.0, 54.5, 50.0, 42.3, 39.6, 37.2, 36.4, 35.7, 35.1, 33.1, 31.6, 31.5, 31.4, 29.8, 29.7, 29.4, 21.3, 21.1, 20.7, 19.4, 16.5, 12.5, 12.1; ESI-HRMS calcd for C31H54O6N [M+NH4]+536.3946, Found 536.393 8。
1.2.7 化合物11的合成 化合物10 (0.025 g, 0.048 mmol),糖基供体3 (168 mg, 0.20 mmol)和分子筛AW 300 MS (100 mg)于反应管中,加入1,2-二氯乙烷(1.2 mL),室温下搅拌1 h,加入(4-MeOPh)3PAuCl(5.7 mg, 0.0096 mmol)和AgB(C6F5)4(15 mg, 0.0192 mmol),40 ℃下反应20 h,TLC检测反应完全。过滤掉分子筛、浓缩,硅胶柱层析(Toluene:Acetone=38:1-Petroleum ether: AcOEt=2.6:1)得到化合物11(0.072 mg, 0.043 mmol, 90%):[α]25D+19.9 (c0.65, CHCl3);1H NMR (500 MHz, CDCl3)δ8.00 (d,J=7.7 Hz, 4H), 7.94 (t,J=7.8 Hz, 4H), 7.92-7.87 (m, 4H), 7.86-7.79 (m, 4H), 7.57-7.46 (m, 6H), 7.45-7.30 (m, 15H), 7.29 (d,J=7.6 Hz, 3H), 5.94-5.83 (m, 2H), 5.72-5.58 (m, 2H), 5.50 (dd,J=16.8, 7.4 Hz,2H), 5.25-5.09 (m, 2H), 4.94 (d,J=7.9 Hz, 1H), 4.81 (d,J=7.8 Hz, 1H), 4.75 (t,J=6.8 Hz, 1H), 4.61 (td,J=11.7, 2.9 Hz, 2H), 4.58-4.45 (m, 2H), 4.16-4.13 (m, 2H), 3.68 (dd,J=9.3, 5.6 Hz, 1H), 3.61-3.49 (m, 1H), 3.38 (dd,J=9.3, 5.6 Hz, 1H), 2.45-2.29 (m, 1H), 2.20-2.10 (m, 2H),2.06-2.01 (m,1H),1.97(s, 3H),1.95 (s, 3H), 0.90 (d,J=8.6 Hz, 3H), 0.89 (s, 3H), 0.81 (s, 3H), 0.76 (d,J=6.7 Hz, 3H);13C NMR (125 MHz, CDCl3)δ170.9, 170.1, 166.1, 166.1, 165.8, 165.8, 165.2, 165.2, 165.1, 165.0, 140.2, 133.4, 133.2, 133.1, 133.0, 129.8, 129.74, 129.72, 129.70, 129.6, 129.57, 129.4, 129.3, 128.84, 128.79, 128.38, 128.35, 128.31, 128.26, 121.7, 101.3, 100.1, 80.2, 75.7, 75.4, 75.0, 73.0, 72.96, 72.13, 72.1, 72.0, 71.9, 70.0, 69.8, 63.3, 63.2, 55.9, 54.4, 49.9, 42.3, 39.5, 38.7, 37.0, 36.5, 35.1, 33.2, 33.1, 31.5, 31.3, 29.7, 29.5, 22.7, 21.3, 21.0, 20.7, 19.2, 16.6, 12.4, 12.1; ESI-HRMS calcd for C99H102O24Na [M + Na]+1697.6653, Found 1697.6621。1.2.8 11-deoxyheloside A 的合成 化合物11 (36 mg, 0.021 mmol)和NaOH (63 mg, 1.53 mmol)溶于THF和MeOH (7 mL, v:v=1:1)中,50 ℃下反应18 h,用Dowex-50(H+)树脂调pH至中性。过滤、浓缩,硅胶柱层析[CH2Cl2:MeOH (8% H2O)=1:1]得到11-deoxyheloside A (16 mg, 0.0205 mmol, 97%):[α]25D-27.9 (c0.55, MeOH) [lit.2-28.1 (c0.23, MeOH) ];1H NMR (600 MHz, Pyridine-d5/D2O~9:1)δ5.37 (d,J=2.4 Hz, 1H), 5.06 (d,J=7.4 Hz, 1H), 4.83-4.80 (m, 2H), 4.55-4.53 (m, 2H), 4.40-4.15 (m, 9H), 4.11-4.00 (m, 4H), 4.00-3.95 (m, 4H), 3.66-3.64 (m, 1H), 2.78-2.76 (m, 1H), 2.54-2.49 (m, 2H), 2.35-2.30 (m, 1H), 2.21-2.18 (m, 1H), 2.15-2.07 (m, 1H), 2.06-1.97 (m, 2H), 1.93-1.85 (m, 1H), 1.85-1.69 (m, 7H), 1.56-1.30 (m, 9H), 1.22 (d,J=6.2 Hz, 3H), 1.12 (s, 3H), 1.02 (d,J=6.3 Hz, 3H), 0.94 (s, 3H), 0.93-0.86 (s, 5H);13C NMR (125 MHz, CD Cl3)δ141.0, 122.0, 104.7, 102.5, 78.4, 78.3, 75.4, 75.2, 75.1, 71.6, 71.5, 62.70, 62.67, 58.0, 54.9, 50.5, 42.6, 40.4, 39.3, 37.5, 37.1, 37.0, 36.0, 34.2, 32.2, 31.9, 31.5, 30.2, 21.1, 19.4, 17.6, 14.9, 13.5; ESI-HRMS calcd for C39H66O14Na [M + Na]+781.4345, Found 781.4334。
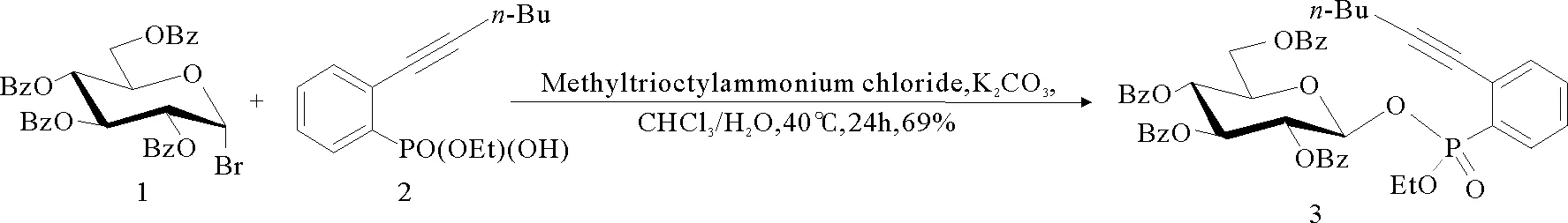
图2 葡萄糖膦酸酯3的合成
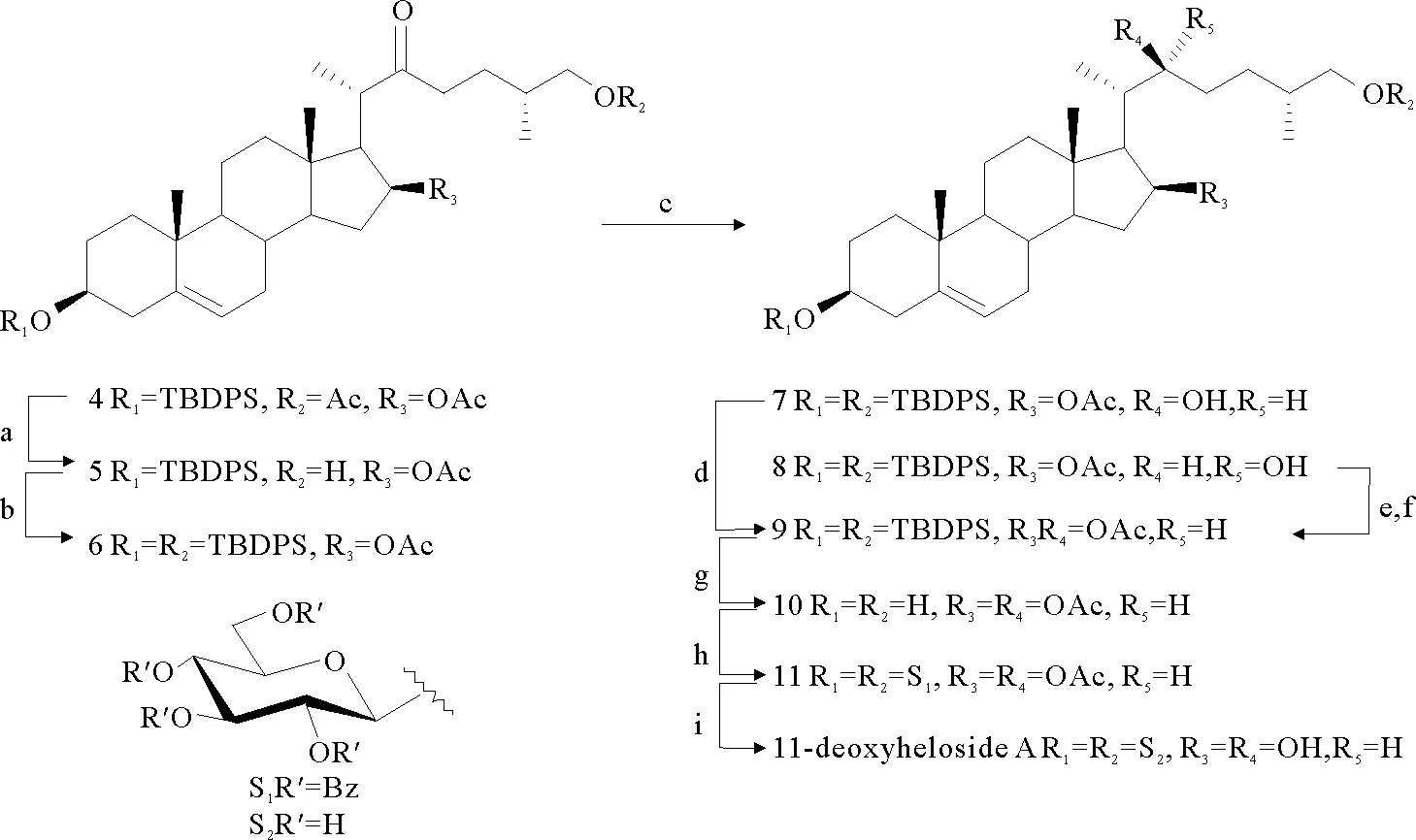
(Reagents and conditions: (a) MeONa/MeOH, r.t., 11.5 h, 87%; (b) TBDPSCl, imidazole, DMF, r.t., 6 h, 90%; (c) NaBH4, CeCl3·7 H2O, THF, 0 ℃-r.t., overnight, 92% (7:8=1:4); (d) Ac2O, Pyridine, r.t., overnight, 92%; (e) MsCl, DMAP, DIPEA, DCM, MS 4Å, -10 ℃, 4 h; (f) DBU-AcOH, toluene, 95 ℃, 11 h, 39% for two steps; (g) TfOH-SiO2(500 mg/ mmol), MeCN/ DCM, 50 ℃, 1 h, 91%; (h) 3, (4-MeOPh)3PAuCl, AgB(C6F5)4, AW 300 MS, 1,2-DCE, 40 ℃, 20 h, 90%; (i) NaOH, THF-MeOH, 50 ℃, 18 h, 97%.)
图3 11-deoxyheloside A的合成
Fig.3 Synthesis of 11-deoxyheloside A
2 结果与讨论
在相转移催化剂作用下,羧酸和酚与溴苷顺利反应形成糖苷键[4]。相转移催化反应可以应用于葡萄糖膦酸酯供体3的制备。以甲基三辛基氯化铵为相转移催化剂,CHCl3和H2O为溶剂(体积比为1:1),葡萄糖溴苷1[5]与膦酸单乙酯2[6]进行SN2反应得到葡萄糖膦酸酯供体3(见图2),产率69%。
苷元部分的合成见图3,化合物4[7]在MeONa/MeOH溶液中选择性脱除26位的乙酰基,得到化合物5。在咪唑的作用下,化合物5与TBDPSCl反应,得到苷元3和26位TBDPS基保护的化合物6,产率为90%。化合物6经Luche还原反应得到两个非对映异构体7和8 (92%, 7:8=1:4)[8]。化合物7的22位羟基为S构型,是期望的还原产物。化合物7发生乙酰化反应得到化合物9,产率为92%。在DIPEA的作用下,化合物8的22R-OH与MsCl反应生成甲基磺酸酯中间体。该中间体在DBU的作用下与AcO-进行SN2反应得到化合物9[9],两步反应的产率为39%。四丁基氟化铵(TBAF)溶液脱除化合物9上的TBDPS基得到化合物10[10],产率为79%。但是生成少量22位乙酰基脱除的副产物,分析是体系中碱性过强导致碱性敏感基团酯键的断裂。加入等当量的醋酸中和体系中的碱性[11],产率没有提高。氢氟酸-吡啶溶液脱除化合物9上的TBDPS基时,产物复杂[12]。最终用三氟甲磺酸负载硅胶(TfOH-SiO2)成功脱除9上的TBDPS基团得到化合物10[13],产率提高至91%。
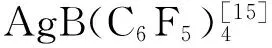
3 结语
本文以胆甾烷化合物4和葡萄糖溴苷1为原料,通过金催化的邻炔基膦酸酯的糖苷化反应,首次完成了胆甾烷型皂苷11-deoxyheloside A的合成,为合成黄地百合中系列胆甾烷型皂苷奠定了基础。
[1] Hostettmann K, Marston A.Saponins [M]. New York: Cambridge Univercity Press, 1995.
[2] Challinor V L, Stuthe J M U, De Voss J J, et al. Structure and bioactivity of steroidal saponins isolated from the roots ofchamaeliriumluteum(False Unicorn) [J]. J Nat Prod, 2012, 75: 1469-1479.
[3] Cheng M S, Wang Q L, Yang Z, et al. Total Synthesis of Methyl Protodioscin: A Potent Agent with Antitumor Activity [J]. J Org chem, 2003, 68: 3658-3662.
[4] Wang P, Li C X, Li Y X, et al. Synthesis of two bidesmosidic ursolic acid saponins bearing a 2, 3-branched trisaccharide residue [J]. Carbohydr Res, 2005, 340: 2086-2096.
[5] Konradsson P, Fraser-Reid B. Conversion of pent-4-enyl glycosides into glycosyl bromides [J]. J Chem Soc, Chem Commun, 1989, 16: 1124-5.
[6] Peng A Y, Zhang X Y, Ding Y X. A Convenient and Applicable Route to Synthesize 2-(1-Alkynyl) phenylphosphonates [J]. Heteroatom Chem, 2005, 6: 529-534.
[7] 彭雁南. 澳洲茄碱和(22S, 25R)-α-茄碱的合成 [D]. 青岛: 中国海洋大学, 2013.
[8] Matsuya Y, Masuda S, Nemoto H, et al. Synthesis and antitumor activity of the estrane analogue of OSW-1 [J]. Eur J Org Chem, 2005, 803-808.
[9] Shi X X, Shen C L, Yao J Z, et al. Inversion of secondary chiral alcohols in toluene with the tunable complex of R3N-R′COOH [J]. Tetrahedron: Asymmetry, 2010, 21: 277-284.
[10] Mukai C, Itoh T, Hanaoka M. New glycosylation reaction based on alkyne-Co2(CO)6complex [J]. Tetrahedron Lett, 1997, 38(26): 4595-4598.
[11] Lam S N, Gervay-Hague J. Solution- and solid-phase oligosaccharide synthesis using glucosyliodides: a comparative study [J]. Carbohydr Res, 2002, 337: 1953-1965.
[12] Li Y, Roy B, Liu X Y. New insight on 2-naphthylmethyl (NAP) ether as a protecting group in carbohydrate synthesis: a divergent approach towards a high-mannose type oligosaccharide library [J]. Chem Commun, 2011, 47:8952-8954.
[13] Yan S Q, Ding N, Li Y X, et al. An efficient and recyclable catalyst for the cleavage oftert-butyldiphenylsilyl ethers [J]. Carbohydr Res, 2012, 354: 6-20.
[14] Zalesskiy S S, Sedykh A E, Ananikov V P, et al. Efficient general procedure to access a diversity of gold(0) particles and gold(I) phosphine complexes from a simple HAuCl4Source.Localization of homogeneous/heterogeneous system′s interface and Field-Emission scanning electron microscopy study [J]. J Am Chem Soc, 2013, 135: 803-808.
[15] Tanaka S, Takashina M, Tokimoto H, et al. Highly β-selective mannosyl-ation towards man 1-4 GlcNNAc synthesis: TMSB(C6F5)4as a Lewis acid/Cation trap catalyst [J]. Synlett, 2005, 15: 2325.
责任编辑 徐 环
Synthesis of 11-deoxyheloside A
ZHANG Xue-Wei, WANG Peng, SONG Ni, ZHANG Xiu-Li, WANG Cong, LI Ming
(The Key Laboratory of Marine Durgs, Ministry of Education, School of Medicine and Pharmacy, Ocean University of China, Qingdao 266003,China)
Synthesis of 11-deoxyheloside A, a natural cholestan saponin, was accomplished for the first time. Glucopyranosyl phosphonate 3 was prepared in 69% yield by treatment of glucopyranosyl bromide 1 with phosphonic acid monoethyl ester 2 under the phase-transfer catalysis conditions. Cholestan 4 was subjected to Luche reduction followed by acetylation and deprotection oftert-butyldiphenylsilyl mediated by triflic acid supported on silica gel to afford aglycone 10. Glycosylation of 10 with glucopyranosyl phosphonate 3 under the promotion of gold(I) catalyst followed by removal of benzoyl protecting groups in the presence of NaOH in MeOH produced 11-deoxyheloside A in 97% yield.
11-deoxyheloside A; glucopyranosyl phosphonate; gold-catalyzed glycosylation
国家自然科学基金面上项目(21272220)资助
2014-03-11;
2014-05-16
张雪薇(1987-),女,硕士生。E-mail:zhangxuewei1314@126.com
** 通讯作者: E-mail:lmsnouc@ouc.edu.cn
R914.5
A
1672-5174(2015)04-072-06
10.16441/j.cnki.hdxb.20140076
