基于最大信息系数的永久性房颤差异表达基因识别
2015-02-21刘汉明饶妮妮罗恒荣
刘汉明 饶妮妮 李 益 罗恒荣 杨 阳 杨 锋
1(电子科技大学生命科学学院,成都 610054)2(赣南师范学院计算机科学学院,江西 赣州 341000)
基于最大信息系数的永久性房颤差异表达基因识别
刘汉明1,2饶妮妮1*李 益1罗恒荣1杨 阳1杨 锋1
1(电子科技大学生命科学学院,成都 610054)2(赣南师范学院计算机科学学院,江西 赣州 341000)
心房颤动是一种常见的、与年龄相关的心律失常,由其所导致的脑卒中具有高致残率和致死率。通过对高通量基因表达谱的分析,可以帮助理解心房颤动的生物学过程和功能紊乱机制,并发现相关致病基因。新型的非参数统计方法——最大信息系数,在探索双变量之间的关联方面具有独特的优点。利用该方法,发现差异与非差异基因表达值与样本表型之间的关联程度不同,构建差异表达基因识别方法。对永久性房颤基因表达谱GSE2240的分析,识别出41个差异表达基因,其中有14基因是已有工作未发现的新差异表达基因。信号通路和富集分析表明,这些差异表达基因与房颤高度相关。同时,对乳腺癌基因表达数据GSE24037的分析,进一步说明该方法在差异表达基因识别方面的有效性。最大信息系数的非参数特性与抗噪能力,使它非常适合于差异表达基因的识别。
最大信息系数;心房颤动;差异表达基因
引言
心房颤动(简称“房颤”)是一种常见的持续性心律失常[1-4]。房颤的发生率随着年龄增长而增加,人群发病比率可从50岁时的0.5%上升到80岁(或以上)时的10%[5-6]。房颤已经被证明是脑卒中的一个独立风险因素,房颤病人脑卒中归因危险度是正常人的3~5倍[7];而且,与非房颤病人相比,由房颤引起的脑卒中后果更为严重,有更高的致残率和致死率[5]。探索房颤的发生和发展机理,是摆在研究人员面前的一个重要课题。
基因表达分析在研究生物体特性和基因功能中起着至关重要的作用[8],它可以不考虑具体的生物条件、细胞周期状态、组织和生物个体而鉴别出差异表达基因。通过基因的差异表达信息,可以精确地洞察生物学过程和功能紊乱,并发现相关的致病基因。有许多基于基因表达的研究,为发现和理解房颤的病理或治疗机制作出了重要贡献。Kim等研究了来自房颤和氧化应激下的人类心肌基因转录谱,为检验氧化应激对房颤的影响,他们使用放射cDNA微阵列来评估1 152个已知基因的表达变化,并在房颤病人中发现了30个上调基因和25下调基因。他们的研究表明,氧化应激的细胞内机制在房颤的发展过程中扮演着关键的角色,并提出一种新型的、可能的治疗方案——对房颤病人进行抗氧化治疗[9]。为获得人类心脏衰竭的分子发病机理,Ohki-Kaneda等人分析了总共17份人类右心肌样本的超过12 000个基因。他们的分析表明,站在基因表达角度,正常人与房颤病人的心肌存在着明显的差异[10]。文献[11]则通过对房颤病人与正常人群的基因表达谱数据的比较分析,找到1 434个差异表达基因(differentially expressed gene,DEG),并发现使用心室状标记的去分化是房颤的一个基本特征[11]。另外,文献[12]采用间接主成分分析OPCA(oblique principal component analysis),对文献[11]的数据重新分析,找到了一些新的与永久性房颤(pmAF,permanent Atrial Fibrillation)相关的差异表达基因,并发现了基因表达中的“吸引子样”属性[12]。
尽管已有的研究在差异基因表达的分析中取得了很好的成绩,但仍或多或少地存在一些局限性。例如,当参考样本的信号强度很小时,可能使倍率法得到很大的倍率,而这个具有很大倍率的点很可能会当作噪声干扰被去除。另外,合适倍数阈值的选择也是该方法的一大难题。t-检验法虽然可以克服倍率法的一些缺点,但为了提高分析精确度,需要足够多的样本,从而导致其花费很大或者实验过程冗长。若减少样本,又会使总体方差被严重低估,而带来较高的假发现率;SAM(significance analysis of microarrays)法则可能丢失一些与房颤有关的差异表达基因[12]。为了进一步拓展基因表达谱的分析手段,引入最大信息系数以构建一种鉴别差异表达基因的新方法,并将此方法用于永久性房颤差异表达基因的识别。
最大信息系数(maximal information coefficient, MIC)是由Reshef等在2011年提出的探索性统计工具,它主要用于探索两个变量之间是否存在某种函数关系[13]。对于基因表达数据,可以把其中的样本表型和某基因表达值看作是两个变量,并使用MIC来探索它们之间的关系,从而推断该基因是否为差异表达基因。通过MIC对永久性房颤基因表达谱数据进行分析,识别了分布在45个探针上的41个差异表达基因,其中14个差异表达基因是已有研究工作未识别的。基于DAVID[14]的信号通路和富集分析表明,MIC所识别的差异表达基因与房颤高度相关。MIC用于差异表达基因识别,获得了与现有工作平均4.5%的结果相同率,而现有工作之间的相同率只有1.9%,这说明MIC方法具有更好的适应性。
1 材料和方法
1.1 材料
研究所用材料为基因表达数据,来源于美国国家生物技术信息中心(National Center for Bio-technology Information,NCBI)的基因表达数据库(Gene Expression Omnibus, GEO),是编号为GSE2240、基于Affymetrix U133A基因芯片的基因表达谱数据。该数据包含35份样本和22 283个基因,其中有10个永久性房颤病人的心房肌样本,20个窦性心律的心房肌样本,5个非心衰的左心室肌样本[11]。考虑到病例/对照样本来源的一致性,笔者从该数据中去除了5个左心室肌样本,把10个永久性房颤病人的心房肌作为疾病组样本,20个窦性心律的心房肌作为对照组样本。此外,还选取另一个GEO数据GSE24037,以进一步验证本研究所采取的方法。这是乳腺癌基因表达数据,包含18个正常样本和18个乳腺癌样本及22 283个基因的表达值。
1.2 方法
作为一个探索性分析工具,MIC可用于探索几百个变量值中可能存在的、重要的、尚未发现的关系,比如存在于大规模的全基因组数据集中基因与疾病之间的关联关系。文献[13]定义双变量集D的MIC为
(1)
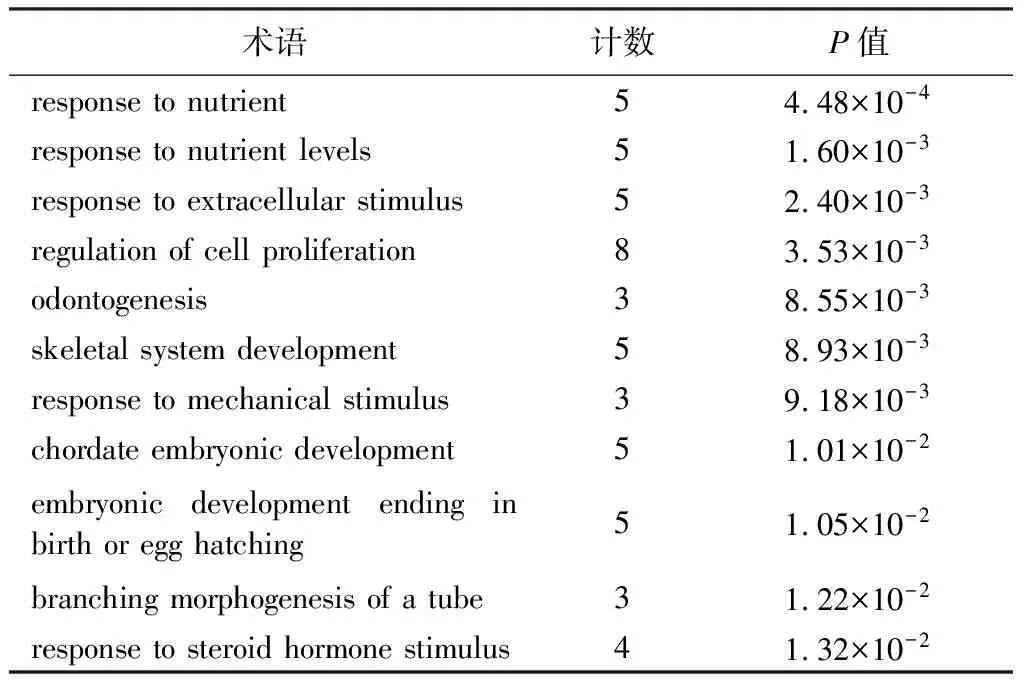
这里,双变量集是指包含有两个变量的数据集(如病例-对照基因表达谱数据中样本的表型和基因的表达值就是两个变量),n表示样本大小,B为把所有数据点映射于二维平面的xy格栅大小的上限(通常,ω(1) (2) 式中,I*(D)表示D中的两个变量之间的互信息。 根据式(1)和式(2),MIC可以理解为一种特殊的、被log(min{x,y})归一化了的最大信息熵,那是因为对于两个变量v1、v2来说,其互信息可表示为 (3) 熵被认为是随机变量不确定性的表征,这意味着熵可用于表示变量间的关系(如文献[15]的工作),即MIC用于探索基因与疾病间的关联关系是可行的。基于式(1)和式(2),MIC的计算被转化为求解max{I*(D)}。为了求解这个最大互信息,DavidN.Reshef等创造性地提出了一种通过探索所有格栅(格栅数量取决于最大分辨率)来计算max{I*(D)}的新颖方法,以达到计算MIC的目的。 该方法的本质是以二维平面上的xy格栅来代替数据集D中的所有点。由于实际使用时xy平面上的格栅数不可能无穷,所以这只是一种近似的方法。不过,笔者的实验表明,这种近似法具有足够的精度。下面详细讨论max{I*(D)}的求解过程。 不失一般性,把数据对(x,y)看作是函数y=f(x)在xy平面上的一系列点。为了计算max I(X;Y),在x、y方向把这些点分成p组和q组(p,q≥2,且pq maxI(X,Y)≈maxI(P;Q)= (4) 为计算方便,先在y方向分成固定的q组,因此H(Q)为常量,那么 maxI(P,Q)|Q= (5) 简便起见,使用Nj表示P中c1~cj所包含点的数量。不失一般性,令F(m,l)=max{H(P)-H(P,Q)}(l为P的第l个分组,l≥2;m=Nl,l (6) 式中,i=Nl-1;H(〈i,m〉,Q)表示x方向第i 个点到第m个点构成的组与y方向所有q个组的联合熵。 这样,只要根据式(6)递推出F(N, p)并代入式(5),即可求得最大互信息maxI(P,Q)|Q。类似地,可以求出maxI(P,Q)|P,因此有 maxI(P,Q)= max{maxI(P,Q)|P,maxI(P,Q)|Q} (7) 如果把双变量数据集看作是二维平面上的一系列点,则max{I*(D)}=maxI(X,Y)。这样,利用式(6)计算F(k,1)(k∈[N2,N]),然后利用该式递推出F(N,p),即可进一步求出max{I*(D)}。显然,p、q值越大,即xy平面所画的格栅越多,分辨率就越高,互信息的计算精确度也越高。不过,高的分辨率会增加计算量,这就是式(1)中要求xy 值得指出的是,MIC模型有一个诱人的特性,这个特性就是随着样本数的增加,无论是非常量无噪函数关系,还是更大的无噪关系类(包含无噪函数关系的重叠),MIC值均会以概率1趋近于1,而对于统计独立变量则会趋于0[13]。这样,用MIC来描述变量间的关联,会使MIC值聚集于[0,1]范围的两端。因此,使用MIC来鉴别变量间是否存在关联,可以提高区分能力。 对于基因表达数据,可以把样本表型和基因表达值看作是两个变量。一个给定的数据集,其表型固定,但各基因间的表达值不同,使得各基因的MIC值不相同。更进一步,由于差异与非差异表达基因的表达值的特性存在较大的不同,所以差异与非差异表达基因的MIC值也会有较大不同。这样,根据基因的MIC值就可以推断差异表达基因。 MIC是非参数统计量,它与描述对象的具体模型无关。对于基因表达数据,样本表型与基因表达值之间目前尚无可靠的模型来表示它们之间的关联,因此MIC非常适合于基因表达数据的研究。 (11) 这样,就可以在不考虑实际的基因表达模型的条件下,通过简单地计算表型T与表达值gj之间的MIC值,就能推断基因j是否为差异表达基因。 2.1 永久性房颤差异表达基因的识别 通常,当皮尔逊系数在0.6~0.8之间时,被认为是强相关。类似于皮尔逊系数,同时考虑到基因数量庞大,为了降低假阳率,在笔者的实验中,把MIC阈值设定为0.75。查MIC-P值表[13],此时P<3.25×10-5(未校正),达10-5量级,可以满足要求。基于此阈值,通过MIC算法,在GSE2240的U133A的基因表达谱中识别出了41个差异表达基因(见表1)。这些基因包含在45个探针中,其中基因TMP3、COG5和MAX等3个基因对应于多个探针。文献[11]和文献[12]使用了与笔者工作相同的基因表达数据。由表1可见,有26个基因与文献[11]的结果相同,有3个基因与文献[12]的结果一致,另外有14个基因(粗体部分)是用笔者所提出的方法新识别的。 表1 MIC识别的永久性房颤差异表达基因 续表1 序号Affymetrix标识基因基因名称MIC值27203542_s_atKLF9Kruppel⁃likefactor90.77328208952_s_atLARP5Laribonucleoproteindomainfamily,member50.77329218425_atRNF216ringfingerprotein2160.76230201151_s_atMBNL1amuscleblind⁃like(Drosophila)0.75731212196_atIL6STainterleukin6signaltransducer(gp130,oncostatinMrecep⁃tor)0.75732221681_s_atDSPPdentinsialophosphoprotein0.75733204260_atCHGBa,bchromograninB(secretogranin1)0.75634205493_s_atDPYSL4adihydropyrimidinase⁃like40.75635219167_atRASL12RAS⁃like,family120.75636205248_atDOPEY2dopeyfamilymember20.75637210198_s_atPLP1aproteolipidprotein10.75638218906_x_atKLC2akinesinlightchain20.75639219321_atMPP5amembraneprotein,palmitoylated5(MAGUKp55subfamilymember5)0.75640221050_s_atGTPBP2GTPbindingprotein20.7564135147_atMCF2LaMCF.2celllinederivedtransformingsequence-like0.756 注:a文献[11]基于同一基因表达数据的结果;b文献[12] 基于同一基因表达数据的结果;粗体字是笔者的方法所识别的基因 Note:adenote the genes identified by reference [11] based on the same data set;bdenote the genes identified by reference [12] based on the same data set; The bold texts show the genes identified by our method 2.2 预测永久性房颤的有关信号通路 通过DAVID来预测筛选得到的41差异表达基因与永久性房颤有关的信号通路,主要为生物学工作者提供探索大量基因背后机制的功能注释工具集[14, 16]。通过DAVID的分析,所识别的永久性房颤差异表达基因中有BMP7和TGFBR2两个基因被卷入到心肌细胞的ALK(activin receptor-like kinase)信号通路中(见表2,有关信号通路图示,参阅文献[17])。 表2 与永久性房颤有关信号通路的基因 2.3 永久性房颤差异表达基因的富集分析 对表达谱芯片数据进行富集分析,为挖掘与研究目标相关的信息并进行生物学专业解释提供了合理的解决方案[18-19]。把获得的差异表达基因通过DAVID做细胞组分(cellular component)、生物学过程(biological process)和分子功能(molecular function)GO(Gene Ontology)富集分析,结果表明:当P≤ 0.05时,在41个差异表达基因中,有25个可以映射到9个细胞组分生物术语(见表3)中,23个可以映射到32个生物学过程术语(见表4)中,3个可以映射到1个分子功能生物术语(见表5)中。 表3 永久性房颤差异表达基因的细胞组分GO富集分析 表4 永久性房颤差异表达基因的生物学过程GO富集分析 续表4 术语计数P值negativeregulationofcellproliferation5136×10-2positiveregulationoftranscriptionfromRNApolymeraseIIpromoter5149×10-2morphogenesisofabranchingstructure3156×10-2embryonicskeletalsystemdevelopment3169×10-2regulationofossification3173×10-2embryonichemopoiesis2231×10-2Golgitoendosometransport2231×10-2responsetoestrogenstimulus3301×10-2pathway⁃restrictedSMADproteinphosphorylation2306×10-2asymmetricproteinlocalization2306×10-2positiveregulationoftranscription,DNA⁃dependent5337×10-2inductionofapoptosisbyextracellularsignals3338×10-2positiveregulationofRNAmetabolicprocess5346×10-2ossification3355×10-2regulationoftranscriptionfromRNApolymeraseIIpromoter6377×10-2intracellularsignalingcascade8391×10-2bonedevelopment3401×10-2tubemorphogenesis3425×10-2smallGTPasemediatedsignaltransduction4437×10-2embryonicmorphogenesis4444×10-2Golgivesicletransport3450×10-2responsetonutrient5448×10-4responsetonutrientlevels5160×10-3responsetoextracellularstimulus5240×10-3regulationofcellproliferation8353×10-3odontogenesis3855×10-3skeletalsystemdevelopment5893×10-3responsetomechanicalstimulus3918×10-3chordateembryonicdevelopment5101×10-2embryonicdevelopmentendinginbirthoregghatching5105×10-2branchingmorphogenesisofatube3122×10-2responsetosteroidhormonestimulus4132×10-2negativeregulationofcellproliferation5136×10-2positiveregulationoftranscriptionfromRNApolymeraseIIpromoter5149×10-2morphogenesisofabranchingstructure3156×10-2embryonicskeletalsystemdevelopment3169×10-2 表5 永久性房颤差异表达基因的分子功能GO富集分析 2.4 乳腺癌差异表达基因的识别 类似于永久性房颤差异表达基因的识别,基于相同的MIC阈值0.75,MIC方法在乳腺癌基因表达数据中识别出了13个差异表达基因(见表6)。通过GO富集分析,在13个差异表达基因中,有10个可以映射到14个不同的生物术语(见表7)中。 差异表达基因分析是根据表型协变量(分类变量)鉴定组间基因表达值自变量之差异表达的数据挖掘手段,为此可以简单地把表型与基因表达值之间的关系用数学模型式(11)表示。基于这一假设,把最大信息系数(MIC)引入此类研究——计算表型与各基因之间的MIC值,并由此推断这些基因是否差异表达。该方法利用了MIC的非参数统计特性,很好地解决了基因表达尚无确定模型这一现实问题。 3.1 方法的优势与不足 文献[11]采用t检验识别差异表达基因。t检验要求数据正态分布,同时表现出较高的假阳性[20-21],因此,有理由相信,文献[11]所识别的1 155个差异表达基因(已去除未注释和重复基因)包含有一定数量的非差异表达基因。文献[12]的方法属于主成分分析。主成分分析是典型的聚类分析方法,而聚类分析对数据的要求是聚类的各组的组内方差较小,组间方差较大。这种要求对基因表达数据来说,显然过于严苛。 MIC是一种非参数统计方法,具有较好的抗噪能力,比常用探索双变量关系的方法具有更好的发现非函数关系的能力,同时它对函数关系具有更好的均一性[13](即不论对何种函数关系,MIC能够给出几乎相同的值)。一般来说,基因表达数据的噪声较高[20],且表型与基因表达值之间的函数关系尚不明确,使得MIC很适合于基因表达数据的分析。 MIC的不足主要体现在它是一种穷举算法,其时间开销要比现有常用方法的时间开销要长。当使用MIC处理超大规模的数据集时,其算法时间是一个必须加以考虑的因素。 3.2 永久性房颤的ALK信号通路 通过MIC方法,笔者在永久性房颤基因表达数据中识别到BMP7、TGFBR2两个基因与BIOCARTA通路[17]的心肌细胞ALK信号通路有关。 在ALK信号通路中,BMP 蛋白作为配体,首先与其具有丝氨酸/苏氨酸激酶活性的II型受体(BMPRII和ActRIIB)结合, 再招募I 型受体(ALK3/BMPRIA、ALK6/BMPRIB 和ALK2/ActRI)并使之磷酸化。磷酸化的BMPRI也具有丝氨酸/苏氨酸激酶活性, 然后再招募效应分子SMAD1/5/8(R-SMADs), 并使R-SMADs C末端磷酸化。磷酸化的R-SMADs 与SMAD4 结合并转运至细胞核内, 在其他转录因子的协同作用下形成转录复合物,结合至靶基因的调控区域, 从而调控靶基因的表达以发挥生物学效应[22],并最终影响心肌细胞的分化。MIC所识别的差异表达基因BMP7通过影响BMP蛋白的表达来影响心肌细胞的分化,而心肌细胞的不正常分化正是房颤的成因之一[23-24]。 表6 MIC识别的乳腺癌差异表达基因 表7 乳腺癌差异表达基因的GO富集分析 无论是小型昆虫还是大型哺乳动物,转化生长因子TGFβ信号控制着多种细胞过程,包括细胞增殖、识别、分化、凋亡和在胚胎发育过程中诱导特定器官的发生等[25-27]。TGFβ把细胞表面的I型、II型受体丝氨酸/苏氨酸激酶结合在一起并与它们绑定,使TGFBR2磷酸化TGFBR1激酶区完成TGFβ信号的跨膜转导[22]。在心肌的ALK信号通路中[17],转导入细胞质中的TGFβ信号通过垫形成效应器的作用,影响着房室垫的发育。事实上,在心血管系统中,TGFβ可诱导血管生成和心肌细胞肥大、钙化及纤维化等生物功能或出现生物学过程[28-29]。心肌细胞肥大、钙化及纤维化均可导致房颤[23, 30-31],这意味着TGFBR2基因是房颤的重要影响因素。 3.3 永久性房颤的GO富集分析 GO富集分析采用基因选择算法,从芯片数据中寻找在两个组别或多个组别中差异表达的基因,它们和样本组别之间有重要的关联[32]。研究表明,房颤与细胞的胞外区/基质有关[33-34]。表3的细胞组分富集结果表明,有近一半(46%)的基因计数(个或次)富集于胞外区/基质;有17.4%的基因计数富集于细胞膜,这些基因的表达可能影响心肌的Na+、K+、Ca2+离子通道,并引起心房的电重构。临床上常用于房颤治疗的是K+通道阻止剂(如索他洛尔、多非利特和伊布利特)[35],说明离子通道的电重构与房颤有关。另外,表4的生物学过程富集显示,基因计数最高的两个生物学过程是“细胞增殖调节”和“胞内信号级联”。从分子水平来考虑,多个房颤相关的心脏组织改变是由不同信号传导系统活动引起的[33, 36-40]。在一个信号传导活动中,当绑定了特定的受体之后,最先到达的信使将激活“胞内信号级联”,并把胞外信使传入到胞内[41],即“胞内信号级联”这个生物学过程与房颤有关。另一个生物学过程“细胞增殖调节”地异常,则可能引起心肌细胞的不正常增殖。有研究表明,心肌细胞的异常增殖是房颤的成因之一[23-24]。表5所示的基因分子功能富集结果,则进一步说明了卷入到ALK心肌细胞信号通路的基因TGFBR2影响着房颤的产生与发展。 另外,笔者还使用MIC分析了乳腺癌基因表达数据,发现了13个差异表达基因。对这些差异表达基因的GO富集分析表明,基因计数最高的是两个有关“非膜结合细胞器”的生物学术语。已有对肾癌的研究发现,癌细胞最显著的特征是非膜结合细胞器在胞浆内非常缺乏[42]。该研究提示,非膜结合细胞器与癌症相关,意味着MIC识别的这些差异表达基因与乳腺癌风险有关。 本研究表明,MIC作为一种具有较好抗噪性能的非参数统计方法,能很好地分析基因表达谱数据。通过对房颤和乳腺癌基因表达数据的分析,证明了MIC在识别差异表达基因上的有效性。 [1] Schotten U, Ausma J, Stellbrink C,etal. Cellular mechanisms of depressed atrial contractility in patients with chronic atrial fibrillation[J]. Circulation, 2001, 103(5):691-698. [2] Nattel S. New ideas about atrial fibrillation 50 years[J]. Nature, 2002, 415(6868):219-226. [3] 涂承媛, 曾衍钧, 李树信. 房颤心电信号的分形维分析[J]. 中国生物医学工程学报, 2005, 24(2):163-166. [4] 刘靖. 心房颤动的治疗进展[J]. 西部医学, 2013, 25(10):1593-1595. [5] Benjamin EJ, Wolf PA, D’Agostino RB,etal. Impact of atrial fibrillation on the risk of death the Framingham Heart Study[J]. Circulation, 1998, 98(10):946-952. [6] Ho KK, Pinsky JL, Kannel WB,etal. The epidemiology of heart failure: the Framingham Study[J]. Journal of the American College of Cardiology, 1993, 22(4s1):A6-A13. [7] Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham study[J]. Stroke, 1991, 22(8):983-988. [8] 李靖, 陈宇光, 孔祥银. 基因表达系列分析技术的新进展[J]. 生物工程学报, 2001, 17(6):613-616. [9] Kim YH, Lee JH, Lim DS,etal. Gene expression profiling of oxidative stress on atrial fibrillation in humans[J]. Experimental and Molecular Medicine, 2003, 35(5):336-349. [10] Ohki-Kaneda R, Ohashi J, Yamamoto K,etal. Cardiac function-related gene expression profiles in human atrial myocytes[J]. Biochemical and Biophysical Research Communications, 2004, 320(4):1328-1336. [11] Barth AS, Merk S, Arnoldi E,etal. Reprogramming of the human atrial transcriptome in permanent atrial fibrillation expression of a ventricular-like genomic signature[J]. Circulation Research, 2005, 96(9):1022-1029. [12] Censi F, Calcagnini G, Bartolini P,etal. A systems biology strategy on differential gene expression data discloses some biological features of atrial fibrillation[J]. PLoS One, 2010, 5(10):e13668. [13] Reshef DN, Reshef YA, Finucane HK,etal. Detecting novel associations in large data sets[J]. Science, 2011, 334(6062):1518-1524. [14] Jiao Xiaoli, Sherman BT, Huang Da-Wei,etal. DAVID-WS: a stateful web service to facilitate gene/protein list analysis[J]. Bioinformatics, 2012, 28(13):1805-1806. [15] Furlanello C, Serafini M, Merler S,etal. Entropy-based gene ranking without selection bias for the predictive classification of microarray data[J]. BMC bioinformatics, 2003, 4(1):54-73. [16] Huang Da-Wei, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists[J]. Nucleic Acids Research, 2009, 37(1):1-13. [17] Nishimura D. BioCarta[J]. Biotech Software & Internet Report: The Computer Software Journal for Scient, 2001, 2(3):117-120. [18] 刘明, 王米渠, 丁维俊, 等. 表达谱芯片数据的基因功能富集分析[J]. 生物医学工程学杂志, 2010, 27(5):1166-1169. [19] 尤元海, 张建中. 基因表达谱芯片的数据挖掘[J]. 中国生物工程杂志, 2009, 29(10):87-91. [20] 蒋定锋, 潘娟娟, 赵耐青. 差异表达基因筛选方法的比较[J]. 中国卫生统计, 2006, 23(5):417-420. [21] 李运明, 曹文君, 陈长生. 六种基因表达谱数据筛选差异表达基因方法的比较[J]. 中国卫生统计, 2009,(3):250-254. [22] Shi Yigong, Massagué J. Mechanisms of TGF-β signaling from cell membrane to the nucleus[J]. Cell, 2003, 113(6):685-700. [23] Camm AJ, Kirchhof P, Lip GY,etal. Guidelines for the management of atrial fibrillation: The Task Force for the Management of Atrial Fibrillation of the European Society of Cardiology (ESC)[J]. European Heart Journal, 2010, 31(19):2369-2429. [24] Burstein B, Nattel S. Atrial fibrosis: mechanisms and clinical relevance in atrial fibrillation[J]. Journal of the American College of Cardiology, 2008, 51(8):802-809. [25] Patterson GI, Padgett RW. TGFβ-related pathways: roles in Caenorhabditis elegans development[J]. Trends in Genetics, 2000, 16(1):27-33. [26] Dijke PT, Goumans MJ, Itoh F,etal. Regulation of cell proliferation by Smad proteins[J]. Journal of Cellular Physiology, 2002, 191(1):1-16. [27] Massagué J. How cells read TGF-β signals[J]. Nature Reviews Molecular Cell Biology, 2000, 1(3):169-178. [28] Ruiz-Ortega M, Rodríguez-Vita J, Sanchez-Lopez E,etal. TGF-β signaling in vascular fibrosis[J]. Cardiovascular Research, 2007, 74(2):196-206. [29] Clark-Greuel JN, Connolly JM, Sorichillo E,etal. Transforming growth factor-β1 mechanisms in aortic valve calcification: increased alkaline phosphatase and related events[J]. The Annals of Thoracic Surgery, 2007, 83(3):946-953. [30] Shiomi T, Tsutsui H, Hayashidani S,etal. Pioglitazone, a peroxisome proliferator-activated receptor-γ agonist, attenuates left ventricular remodeling and failure after experimental myocardial infarction[J]. Circulation, 2002, 106(24):3126-3132. [31] Everett TH, Olgin JE. Atrial fibrosis and the mechanisms of atrial fibrillation[J]. Heart Rhythm, 2007, 4(3):S24-S27. [32] 马宁, 张正国. 一种基于 Gene Ontology 注释信息的基因选择算法[J]. 中国生物医学工程学报, 2009, 28(5):696-700. [33] Goette A, Staack T, Röcken C,etal. Increased expression of extracellular signal-regulated kinase and angiotensin-converting enzyme in human atria during atrial fibrillation[J]. Journal of the American College of Cardiology, 2000, 35(6):1669-1677. [34] Xu Jun, Cui Guanggen, Esmailian F,etal. Atrial extracellular matrix remodeling and the maintenance of atrial fibrillation[J]. Circulation, 2004, 109(3):363-368. [35] Van-Wagoner DR , Pond AL, McCarthy PM,etal. Outward K+ current densities and Kv1. 5 expression are reduced in chronic human atrial fibrillation[J]. Circulation Research, 1997, 80(6):772-781. [36] Aimé-Sempé C, Folliguet T, Rücker-Martin C,etal. Myocardial cell death in fibrillating and dilated human right atria[J]. Journal of the American College of Cardiology, 1999, 34(5):1577-1586. [37] Li Duosheng, Cardin S, Han Hao,etal. Signal transduction underlying arrhythmogenic atrial remodeling due to congestive heart failure[C] // McCann J, eds. Circulation Conference 2000. Philadelphia: Lippincott Williams & Wilkins, 2000: 153-153. [38] Goette A, Arndt M, Röcken C,etal. Regulation of angiotensin II receptor subtypes during atrial fibrillation in humans[J]. Circulation, 2000, 101(23):2678-2681. [39] Lendeckel U, Arndt M, Wrenger S,etal. Expression and activity of ectopeptidases in fibrillating human atria[J]. Journal of Molecular and Cellular Cardiology, 2001, 33(6):1273-1281. [40] Tuinenburg AE, Van-Veldhuisen DJ , Boomsma F ,etal. Comparison of plasma neurohormones in congestive heart failure patients with atrial fibrillation versus patients with sinus rhythm[J]. The American Journal of Cardiology, 1998, 81(10):1207-1210. [41] Goette A, Lendeckel U, Klein HU. Signal transduction systems and atrial fibrillation[J]. Cardiovascular Research, 2002, 54(2):247-258. [42] Li Maomi, Vuolo MA, Weidenheim KM,etal. Collecting-duct carcinoma of the kidney with prominent signet ring cell features[J]. Modern Pathology, 2001, 14(6):623-628. Maximal Information Coefficient on Identifying Differentially Expressed Genes of Permanent Atrial Fibrillation Liu Hanming1,2Rao Nini1*Li Yi1Luo Hengrong1Yang Yang1Yang Feng1 1(SchoolofLifeScienceandTechnology,UniversityofElectronicScienceandTechnologyofChina,Chengdu610054,China)2(SchoolofMathematicsandComputerScience,GannanNormalUniversity,Ganzhou341000,Jiangxi,China) The atrial fibrillation is a common arrhythmia disease. It usually causes stroke resulting in high risk on morbidity and mortality. It can be insight into the biological processes, the functional disorders of atrial fibrillation and the genes associated with disease risks by analyzing a microarray data. A proposed novel statistical method named as maximal information coefficient (MIC) has excellent performance in exploring the relationship between two-variables. Based on the degree of relationship between the phenotypes and differentially or undifferentially expressed genes, the statistical method was introduced into the analysis of a microarray of permanent atrial fibrillations (GSE2240). Total of 41 genes were identified by the method, in which 14 genes are new differentially expressed genes. The differentially expressed genes identified by MIC were demonstrated that they were related with atrial fibrillation diseases, via the analyses of signaling pathway and enrichment in DAVID. Furthermore, MIC was used to analyze a microarray of breast cancer (GSE24037) to test the method. MIC is a non-parametric statistical method with anti-noise, which leads it to be ideally suit for identifying differentially expressed genes. maximal information coefficient; atrial fibrillation; differentially expressed gene 10.3969/j.issn.0258-8021. 2015. 01.002 2014-07-16, 录用日期:2014-11-04 国家自然科学基金(81171411) Q33 A 0258-8021(2015) 01-0008-09 *通信作者(Corresponding author),E-mail: raonn@uestc.edu.cn

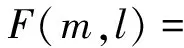

2 结果






3 讨论与结论


