HPLC-MS/MS法测定人血浆头孢克肟浓度的不确定度分析
2015-01-05李力翟学佳
李力,翟学佳
(华中科技大学同济医学院附属协和医院药剂科,武汉 430022)
HPLC-MS/MS法测定人血浆头孢克肟浓度的不确定度分析
李力,翟学佳
(华中科技大学同济医学院附属协和医院药剂科,武汉 430022)
目的 探讨高效液相色谱-质谱联用(HPLC-MS/MS)法测定人血浆头孢克肟浓度的不确定度。方法 对HPLC-MS/MS法测定血浆头孢克肟浓度的全过程进行分析,包括测定重复性、称量、标准溶液的配制、血浆样本的处理、仪器、标准曲线拟合等,计算合成不确定度和扩展不确定度。结果 置信概率为95%时,血浆低(80 ng·mL-1)、中(560 ng·mL-1)、高(9 600 ng·mL-1)浓度头孢克肟的扩展不确定度分别为14.93,23.65,137.95 ng·mL-1。结论 HPLC-MS/MS法测定血浆头孢克肟浓度含量的不确定度主要由标准曲线的拟合、重复性及仪器允差引入。
头孢克肟;高效液相色谱-质谱联用法;血浆;不确定度
测量不确定度是目前国际社会普遍接受和推荐使用的定量说明测量结果质量的一个参数,用于表征合理赋予被测量值的分散性;广义上是指对测量结果正确性的可疑程度[1]。在生物样本分析测试领域,测量不确定度与实验室的质量保证和质量控制密切相关[2-3],其分析结果的可靠性直接决定了药物安全性及有效性的评定,为此,有必要探讨生物样品检测结果的不确定度。
笔者根据国家计量技术规范和不确定度评定的相关资料,结合已发表的文献[4-9],对高效液相色谱-质谱联用(high performance liquid chromatography-tandem mass spectrometry,HPLC-MS/MS)法测定血浆头孢克肟浓度的不确定度进行分析,找出影响不确定度的因素,对不确定度进行评估,最终得出测定结果扩展不确定度值,如实反映测量的置信度和准确度,对实验结果的可信性、可比性和可接受性有非常重要的意义。
1 试药与仪器
1.1 试药 头孢克肟对照品(浙江食品药品检验研究院,含量:87.4%,批号:090831);内标阿莫西林对照品(浙江食品药品检验研究院,含量:86.9%,批号:20100607);甲醇、乙腈,色谱纯(美国Merck公司);水为纯化水;空白血浆来源于武汉市血液中心。
1.2 仪器 Agilent 1200型液相色谱系统:四元梯度泵、在线脱气机、自动进样器、柱温箱(美国Agilent公司);MS/MS系统:API 4000型三重四极杆串联质谱仪,配备电喷雾电离源(Turbo Ionspray),美国Applied Biosystems公司;岛津AUW-220D十万分之一电子分析天平;混合器:XW-80A微型涡旋混合仪(上海沪西分析仪器厂);离心机:Thermo BR4i型高速离心机(法国Thermo公司)。实验室用25 和10 mL量瓶,5 mL单标线移液管和5 mL分度吸量管(玻璃量器均为A级)。
2 方法与结果
2.1 色谱/质谱条件 色谱条件:色谱柱:Ultimate C18柱(150 mm × 2.1 mm,5.0 μm);流动相:乙腈:20 mmol·L-1乙酸胺(含0.3%甲酸)溶液=22:78;流速:0.35 mL·min-1;柱温:25 ℃;自动进样箱温度:4 ℃。
质谱条件:离子检测方式采用多反应离子检测;离子极性:正离子;离子化方式:气动辅助电喷雾离子化;电喷雾电压 :5 500 V;离子源温度 :500 ℃;辅助气1压力 :344.75 kPa,辅助气2压力:344.75 kPa;气帘气压力:172.38 kPa;碰撞气:41.37 kPa;入口电压:10 V;出口电压:10 V。头孢克肟和内标阿莫西林定量分析离子对以及各自的检测条件见表1。
2.2 溶液的配制
2.2.1 头孢克肟标准系列溶液的配制 精密称取盐酸头孢克肟对照品11.44 mg(含头孢克肟10 mg)置50 mL量瓶,用流动相溶解并稀释至刻度,配得200 μg·mL-1头孢克肟母液。分别用流动相依次稀释配制成含头孢克肟120 000(C1),40 000(C2),15 000(C3),5 600(C4),2 400(C5),800(C6),300(C7)ng·mL-1的系列标准曲线溶液和含头孢克肟800(低),5 600(中),96 000(高)ng·mL-1的质控标准溶液。溶液配制过程中需用到10 mL量瓶、5 mL单标线移液管和5 mL分度吸量管。
2.2.2 内标溶液的配制 精密称取阿莫西林对照品11.51 mg(含阿莫西林10 mg),置于10 mL量瓶中,用甲醇溶解并定容,得到1.0 mg·mL-1阿莫西林储备液,置于4 ℃冰箱保存。用5 mL移液管取阿莫西林储备液4 mL定容至100 mL,得40 μg·mL-1的内标工作液。以上各储备液及标准系列溶液均用经过校正的量瓶配制,在4 ℃冰箱内保存备用。
2.3 血浆样品处理 精密吸取血浆样本200 μL,置于1.5 mL Ep管中,精密加入内标溶液20 μL,涡旋10 s后,加入甲醇0.6 mL,涡旋1 min,然后于21 100×g离心10 min,吸取上清液置于进样瓶中。
2.4 测定和计算 取“2.3”项处理后的上清液5 μL进行HPLC-MS/MS 分析,测定计算头孢克肟的峰面积As和内标峰面积Ai的比值R(R=As/Ai)。以峰面积比值(R)对血药浓度(C)进行加权最小二乘法线性拟合,得标准曲线回归方程。样品用同样的方法分析后,代入回归方程,计算待测血浆中头孢克肟浓度。数据采集分析使用Analyst 1.5.1版软件(美国Applied Biosystems公司)。
2.5 校准 仪器在使用前用标准物质聚丙二醇进行一次校准,表明仪器运行正常。分析样品时,每天做随行标准曲线进行校准。
2.6 数学模型的建立
标准曲线公式:R=a×C+b
血浆中头孢克肟浓度计算公式:C=(R-b)/a
其中:a为标准曲线回归方程斜率,b为标准曲线回归方程截距,R为头孢克肟与内标峰面积之比,C为头孢克肟最终血浆测得浓度。
2.7 不确定度分析
2.7.1 分析不确定度分量 根据实际前处理方法,不确定度来源主要有操作重复性、天平称量、量瓶、移液管、标准溶液配制、标曲拟合、HPLC-MS/MS仪器等因素,上述影响见图1。
2.7.2 测定结果的A类不确定度评定 重复性引起的不确定度用A类评定程序。平行配制浓度为浓度为80,560,9 600 ng·mL-1的质控样本各5份,共配制3批,结果见表2。
一次测量的标准偏差的贝塞尔公式为:

S1(x,L)=1.966 ng·mL-1
S2(x,L)=2.022 ng·mL-1
S3(x,L)=1.334 ng·mL-1
低浓度(L)15次测定结果的标准不确定度为:
表1 头孢克肟和内标的检测条件


试药监控离子对(相对分子质量)采集时间/ms去簇电压/V碰撞能量/eV保留时间/min头孢克肟454.1/285.0520060212.06±0.3阿莫西林366.05/249.0520060152.40±0.3

图1 不确定度来源分析图
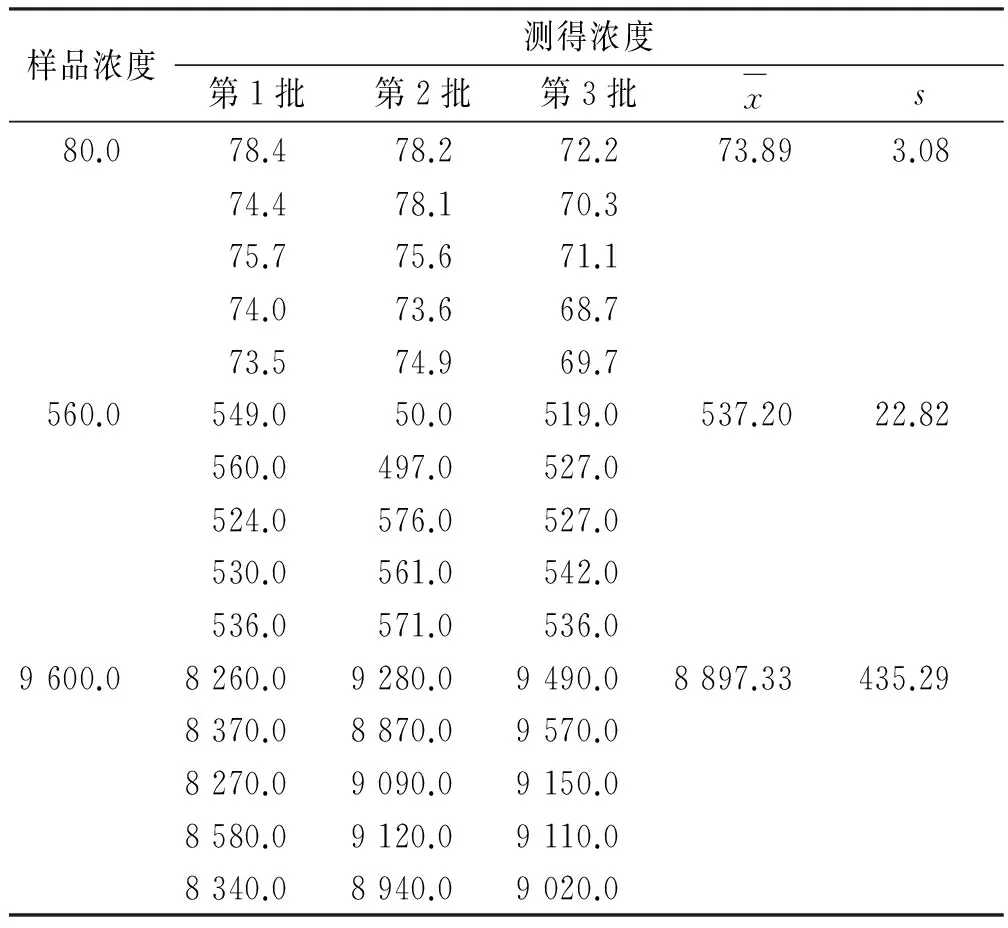
表2 头孢克肟样品重复测定结果Tab.2 Determination results on the repeatability of cefixime detection ng·mL-1
测定结果的相对标准不确定度为:
同法计算中浓度和高浓度相对标准不确定度为:Urel(1,M)=0.016 0;Urel(1,H)=0.003 9。
2.7.3 测定结果的B类不确定度评定
2.7.3.1 天平称量头孢克肟及内标时引入的不确定度 采用B类评定。用十万分之一电子天平,可自动减皮重。
数学模型y=m+△0+△
其中m为透明部分,△0为由自动调零引起的误差,△为天平的非线性误差。

自动调零作为一次扣皮重,其a0=a,则自动调零引起的标准测量不确定度为:U(△0)=a0/k0=0.002 9 mg
不考虑重复性误差时,天平的标准不确定度:
Uc(x)=0.050 mg
头孢克肟和内标称量相对标准测量不确定度分别为(x1,x2分别为头孢克肟和内标称量值):
Urel(x1)=Uc/x1=0.003/10=0.000 3
Urel(x2)=Uc/x2=0.003/10=0.000 3
2.7.3.2 标准溶液配制引入的测量不确定度 采用B类评定程序。配制储备液及标准溶液中用到的量瓶容积为100 mL,其允差为0.1 mL,25 mL量瓶允差为0.03 mL。5 mL移液管和10 mL移液管允差分别为0.015和0.020 mL。
Urel(3)=
2.7.3.3 样品不均匀性引起的测量不确定度 用B类评定程序,因为样品均为液态,并充分涡旋混合,可忽略不计由此带来的不确定度。
2.7.3.4 标准曲线拟合过程中引入的不确定度 采用B类评定程序。 以待测物浓度(X)为横坐标,待测物与内标的峰面积比值(Y)为纵坐标,采用加权(权重为1/X2)最小二乘法进行线性回归,共拟和3条标准曲线,斜率和截距见表3。

表3 各拟合标准曲线的参数Tab.3 Parameters of calibration curves
共3条标准曲线,7种浓度,共测定21次。
Y=a+bX,误差方程为:vi=yi-(axi+b)
∑vi2=∑[yi-(axi+b)]2
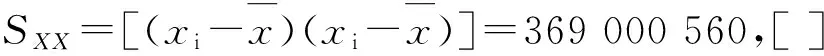
按前述样品处理方法处理高、中、低3种浓度质控样品各15个,平均浓度:XL=73.89 ng·mL-1,XM=537.20 ng·mL-1,XH=8 897.33 ng·mL-1。标准测量不确定度为:
P为测定X的总次数(P=15)
N为测定对照品血浆的总次数(N=3×7=21)
a为标准曲线斜率
b为标准曲线截距
标准曲线对于高、中、低3种浓度的相对标准测量不确定度为:
低浓度:Urel,L(4)=7.45/73.89=0.100 8
中浓度:Urel,M(4)=7.28/537.20=0.013 6
高浓度:Urel,H(4)=9.52/8 897.33=0.001 1
2.7.3.5 仪器测量不确定度 所用仪器为API 4000型三重四极杆串联质谱仪,其定量允差为1%,其相对标准不确定度为:
2.7.3.6 温度引起的相对不确定度 由于药物和内标都是在相同的温度环境下测定的,因此温度对浓度的影响引起的不确定度可以忽略不计。
2.7.3.7 对照品含量引起的测量不确定度 对照品未提供不确定度,视为真实值,引入不确定度为零。
2.8 各不确定度分量的统计直方图 见图2~4。
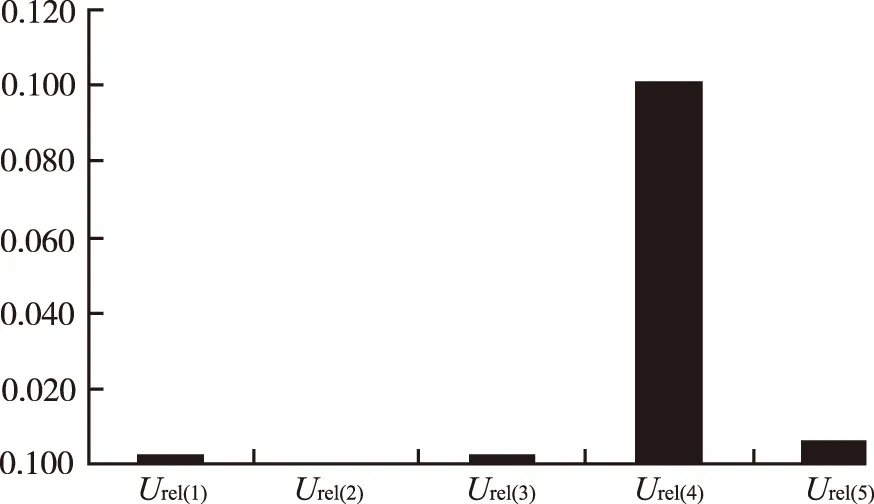
图2 低浓度不确定度分量的统计直方图
Fig.2 Histogram of uncertainty components at low concentration

图3 中浓度不确定度分量的统计直方图
Fig.3 Histogram of uncertainty components at middle concentration
2.9 标准不确定度的合成 根据不确定度传播率对以上各相对不确定度进行合成:
低、中、高浓度各自的合成相对标准测量不确定度
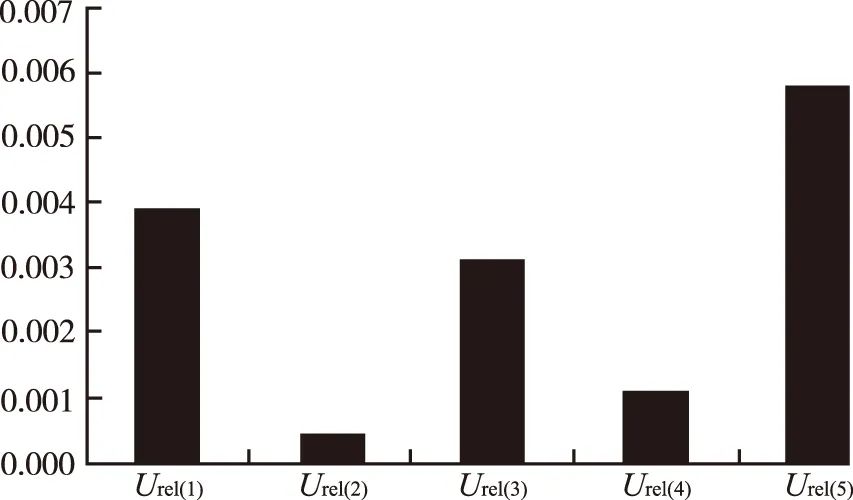
图4 高浓度不确定度分量的统计直方图
Fig.4 Histogram of uncertainty components at high concentration
分别为:Uc,rel,L=0.101 1,Uc,rel,M=0.022 0,Uc,rel,H=0.007 5。
3种浓度测定的合成标准测量不确定度分别为:Uc,L=0.1011×73.89=7.47 ng·mL-1,Uc,M=0.022 0×537.20=11.83 ng·mL-1,Uc,H=0.00 75×8 897.33=68.98 ng·mL-1。
扩展不确定度,取k=2,P=95%,简易评定:UL=k×7.47=14.93 ng·mL-1,UM=k×11.83=23.65 ng·mL-1,UH=k×68.98=137.95 ng·mL-1。
血浆中头孢克肟不同浓度质控测定结果分别为:低浓度(73.89±14.93) ng·mL-1,中浓度(537.20±23.65) ng·mL-1,高浓度(8 897.33±137.95) ng·mL-1,k=2,P=95%。
3 讨论
血浆中头孢克肟含量的HPLC-MS/MS法测定过程中,分析各测量不确定度分量,线性拟合过程对低质控样本不确定度的贡献最大。对于中、高浓度依次是重复性和仪器测量引起的不确定度。由结果可知,对测定结果产生影响的主要因素为标准曲线的拟合、重复性及仪器允差等因素。在实际操作过程中,需着重注意这些影响因素,并采取相应的措施,将测量结果的不确定度进一步减小。
测定过程中涉及到的样品(标准溶液、血浆)均为液态样品,且充分混匀,样品不均匀度很小,故样品不均匀性引入的不确定度非常小,可忽略此部分的不确定度。实验环境温度相对恒定(控制在约20 ℃),温度对于测定结果的影响较小,可忽略不计。
不确定度对于优化实验方法和控制实验质量具有重要的意义。生物样本基质成分复杂,处理步骤繁琐,干扰因素较多,不确定度评定较为困难。笔者对HPLC-MS/MS法测定血浆中头孢克肟的不确定度进行分析,找出影响不确定度的因素,对不确定度进行评估,如实反映测量的置信度和准确度,为方法学研究提供参考。
[1] 国家质量技术监督局.测量不确定度评定与表示[S].JJF1059-1999.
[2] 施昌彦.测量不确定度评定与表示指南[M].北京:中国计量出版社,2005:32.
[3] 陆明,范国荣,汪杨,等.测量不确定度在药品领域的应用[J].医药导报,2013,32(8):1053-1057.
[4] 严蓓,杨晨,李扬,等.HPLC法测定人血浆中奥硝唑浓度的不确定度评定[J].药物分析杂志,2011,31(9):1797-1803.
[5] 杜慧,邢逞,史永强,等.利巴韦林纯度标准物质的定值及不确定度评估研究[J].医药导报,2014,33(6):779-784.
[6] 程颖,李涛,王洪韵,等.重铬酸钾滴定液浓度的不确定度分析[J].中国现代应用药学,2014,31(1):90-92.
[7] 罗淑青,周征.气相内标法测定甘氨双唑钠中乙醇残留量的不确定度评定[J].中国现代应用药学,2015,32(2):173-177.
[8] 秦立,王莹,张利,等.HPLC测定氯氮平片含量的测量不确定度评定[J].中国现代应用药学,2015,32(2):189-194.
[9] 张现化,杨毅恒,杨丽,等.液质联用法测定人血浆中利培酮浓度的不确定度评定[J].药物分析杂志,2010,30(3):366-371.
Uncertainty of Measurement for Cefixime in Human Plasma by HPLC-MS/MS
LI Li,ZHAI Xuejia
(DepartmentofPharmacy,UnionHospital,TongjiMedicalCollege,HuazhongUniversityofScienceandTechnology,Wuhan430022,China)
Objective To develop a method on evaluating the uncertainty during the determination of cefixime in human plasma by high performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS). Methods We analyzed various factors that can cause the uncertainty in the whole process of determination,which includ repeatability,weighting,preparation of standard solutions,sample preparation,equipment error and calibration curve fitting.The expanded uncertainty was evaluated by calculating uncertainty of each component and combined. Results The expanded uncertainty for low (80 ng·mL-1),medium (560 ng·mL-1) and high (9 600 ng·mL-1) level of cefixime was 14.93,23.65,137.95 ng·mL-1,respectively (P=95%). Conclusion The uncertainty of determining cefixime in human plasma by HPLC-MS/MS is mainly caused by the calibration curve fitting,repeatability and HPLC-MS/MS error.
Cefixime; High performance liquid chromatography-tandem mass spectrometry;Plasma; Uncertainty
2014-04-04
2014-10-10
李力(1982-),女,湖北襄阳人,药师,硕士,研究方向:临床药理。电话:027-85726073,E-mail:dsywp@163.com。
翟学佳(1983-),女,湖北武汉人,主管药师,硕士,研究方向:临床药理和药物分析。电话:027-85726073,E-mail:zhaixuejia@163.com。
R978.11;R969.1
A
1004-0781(2015)09-1150-05
10.3870/j.issn.1004-0781.2015.09.007
