基于第二代测序数据识别肿瘤基因突变的工具比较
2014-11-14李文杰孙之荣
李文杰,孙之荣
(清华大学生命科学学院,教育部生物信息学重点实验室,北京100084)
癌症(即恶性肿瘤)一直是导致人类死亡的重要原因之一,也一直是世界范围内的科学家正在攻克的焦点,科学家们一直在探寻其发病原因和有效治疗方法。虽然癌症的病因一般可归纳为原癌基因和抑癌基因中的基因组突变,但具体的突变因病人不同和发生组织不同而不同,所以发现某个病人的致癌突变很有挑战性。
肿瘤发生过程中出现的几类基因组突变包括:点突变(如单碱基替换、碱基插入、碱基删除),拷贝数变化(Copy number variation,CNV),染色体结构变化(如基因融合)。第二代测序技术(Next Genera-tion Sequencing,NGS)作为一种新的实验技术,给癌症研究带来了很多重要的发现。在比较肿瘤和正常样品的测序数据过程中,最重要的是高正确率和高特异性地找到点突变。
因其低成本性和高通量性,第二代测序技术正被研究者使用,以期找到癌症的起因。每次测序实验产生数以百万计的短read,每个位点的碱基都有测序质量。通过比较一个病人的正常和肿瘤组织样品的基因组测序数据,我们可较容易地确定肿瘤组织中发生了哪些点突变。但是样品中细胞纯度、测序错误、碱基测序质量、read比对(与参考基因组比对),都给这个任务带来一定的挑战,特别是对设计插入和删除的点突变。
许多现有的工具先过滤掉一些read和碱基,然后计算报导位点各allele序列的read数目,之后比较位点在正常和肿瘤样品中各allele的read频率。但是这些工具的验证正确率一般都只在54%左右[1],不同工具间的一致性都比较低[2]。
最多被使用的工具有 VarScan[3],SAMtools[4]和MuTect[5]。此文将这些工具应用在一个肺癌病人的血液和癌转移组织的基因组测序数据上,分析它们结果的合理性。同时,此文还获取了该病例的癌转移组织的转录组测序数据,并据此通过它来验证各个工具发现的各个突变位点,以获得工具的验证正确率。
1 数据和方法
此文选择一个肺癌病人的血液组织作为正常组织,癌转移作为肿瘤组织(从肺转移至肝,肺部的原癌组织数据质量很差)[6]。此文应用Bowtie2工具,将测序产生的paired-end read比对到人类参考基因组上(hg19),然后利用SAMtools工具的部分命令,生成按比对位置排序的BAM文件[7]。
对于各个工具,此文简要描述它使用的方法,然后在分析上述样品测序数据时,使用默认的各阈值和推荐的步骤。各工具比较两个样品的全基因组测序数据,以确定发生了点突变的基因组位点。
本文对各突变位点判断肿瘤特异的等位序列,并用病人癌转移组织的转录组测序(RNA-Seq)数据判断这个序列的真实存在性。此方法只验证了被转录了的肿瘤特异等位序列,但也可以作为工具正确率的一种衡量。
2 结果
2.1 VarScan2 工具分析
VarScan2使用各种过滤方法,以得到各位点在样品中的基因型,并计算各等位序列的read频率。此工具只支持最多具有两种等位序列的基因型,一个与参考基因组相同,一个为变异序列;同时这个变异序列在两个样品中需要相同。然后此工具比较两个样品在该位点各等位序列的read频率,并通过Fisher’s Exact检验得到差异显著性水平 p值。VarScan2工具将突变位点分为三种突变状态:Somatic突变、Germline突变、LOH突变;并从三种状态的位点,得到’High-Confidence’位点,各种状态的High-Confidence位点数目见表1。

表1 VarScan发现的各状态突变位点数目Table 1 Number of mutation sites identified by VarScan
但是此工具识别出过多的单碱基替换突变位点,结果中存在比较高的假阳性率,也让工具使用者比较难判断真正的突变位点。对于Somatic状态的突变位点,它在正常样品中的基因型需要是纯合的跟参考基因组一样(R/R,见表2和表3)。对于14 087个位点,在两个样品中有相同的杂合基因型,但是allele的read频率有差异,这些位点并认为是‘LOH’突变,而不是‘Somatic’突变状态(见表2)。
此工具发现的单碱基替换突变位点中,22 207个位点有肿瘤特异的等位序列,4 108个被RNA-Seq数据覆盖,1 359个位点的肿瘤特异等位序列在RNA-Seq数据中存在(验证正确率33%);此工具发现的涉及插入/删除突变位点中,2 710个位点有肿瘤特异的等位序列,422个被RNA-Seq数据覆盖,159个位点的肿瘤特异等位序列在RNA-Seq数据中存在,验证正确率38%。

表2 VarScan发现的单碱基替换突变位点在样品中的基因型统计Table 2 The genotype of SNV sites in both samples

表3 VarScan发现的插入/删除突变位点在样品中的基因型统计Table 3 The genotype of indel sites in both samples
2.2 SAMtools工具分析
SAMtools能利用正常和肿瘤样品的BAM文件,来确定点突变位点。此工具对每个突变位点计算一个CLR值(在输出的VCF文件中),以表明位点在两个样品间的差异显著性大小,值越大表明越显著,范围在0~255(见图1)。此文使用70为阈值,只保留差异显著性大的突变位点,1 012个单碱基替换突变位点,2 578个涉及插入/删除的突变位点。对于涉及插入/删除的突变,此工具在相邻的位点输出相似的序列,所以导致过多的荣誉突变位点。虽然此工具支持一个位点有多种等位序列,但位点的基因型只支持两种等位序列。
此工具发现的位点中,627个单碱基替换突变位点有肿瘤特异等位序列,95个被RNA-Seq数据覆盖,43个位点的肿瘤特异等位序列在RNA-Seq数据中存在,验证正确率45%;1 575个涉及插入/删除的突变位点有肿瘤特异等位序列,186个被RNASeq数据覆盖,105个位点的特异等位序列在RNASeq数据中存在,验证正确率56%。

图1 SAMtools计算的CLR值在突变位点中的分布Fig.1 Distribution of CLR value among mutation sites calculated by SAMtools
2.3 MuTect工具分析
MuTect不支持涉及插入/删除的突变,它过滤掉比对质量低的read,过滤掉覆盖read数低的位点,最后过滤掉有read比对时链偏好性的位点。此工具对每个位点推断发生了突变的可能性:肿瘤样品在此位点与参考基因组序列不同,正常样品没有变异等位序列。对于每个突变位点,此工具还计算其为真实突变的可能性:变异等位序列是真实的,而不是测序错误,然后经过log10转换得到的值。大多数的突变位点的这个可能性值不太高(见图2)。本文以15为阈值,得到输出6 679个单碱基替换突变位点。
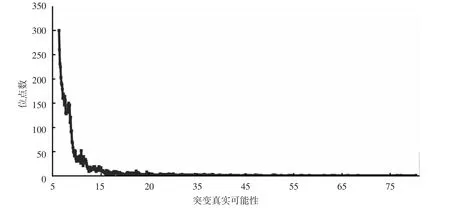
图2 MuTect计算的突变真实可能性在突变位点中的分布Fig.2 Distribution of t_log_fstar among mutation sites calculated by MuTect
此工具发现的单碱基替换突变位点中,5 397个位点有肿瘤特异等位序列,851个被RNA-Seq数据覆盖,305个位点的肿瘤特异等位序列在RNA-Seq数据中存在,验证正确率36%。
3 讨论及结论
现有的工具假定read中测序质量较低的碱基是测序错误,然后将它们过滤掉。这样的方法可能使得结果对选择的阈值敏感。同时,上述工具只在一个位点只支持最多两种等位序列,这可能丢失了肿瘤样品中出现的很多突变位点,原因在于肿瘤细胞中,基因组一个片段可能会出现多个拷贝,每个拷贝可能经过不同的突变过程,这样对应到参考基因组上的某个位点后,可能存在多种等位序列。
各个工具的结果一致性很低,很少比例的位点同时被两个工具发现。这个低一致性可能来自于:(1)它们过滤read和碱基的方式;(2)它们比较两个样品的方法。建议用户尝试多个方法,并用更精确的测序方法验证各自的结果,然后挑选一个有最高验证正确率的工具来使用。
除了此文中讨论的点突变之外,肿瘤组织中还发生了其它类型的基因组突变,如染色体重组。这些突变可能涉及大范围内的碱基,也更难发现和验证。完全研究这些基因组突变依然是比较困难的,更何况确定各突变的功能影响。
当前,ANNOVAR是注释突变功能影响的重要工具之一[8],这个工具(及研究者使用的其它工具)都会忽略不会引起蛋白质序列变化的突变。但是改变蛋白质序列并不是基因组突变产生功能影响的唯一途径:改变序列对转录因子或miRNA的亲和性都会显著得影响功能[9]。因此,需要更多的研究来完全注释基因组突变的功能影响。
References)
[1] KENICHI Y,MASASHI S,YUICHI S,et al.Frequent pathway mutations of splicing machinery in myelodysplasia[J].Nature,2012,478:64-69.
[2] MARTIN L,BERNHARD Y,JOS DE G,et al.Confidence-based somatic mutation evaluation and pioritization[J].PLoS Comput Biol,2012,8(9):e1002714.
[3] DANIEL C,QUNYUAN Z,DAVID E,et al.VarScan 2:Somatic mutation and copy number alteration discovery in cancer by exome sequencing[J].Genome Research,2012,450:65.
[4] HENG L,BOB H,ALEC W,et al.The sequence alignment/map(SAM)format and SAMtools[J].Bioinformatics,2009,25:2078 -9.
[5] KRISTIAN C,MICHAEL S,SCOTT L,et al.Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples[J].Nature Biotechnology,2013,410:60.
[6] YOUNG S,WON-CHUL L,THOMAS B,et al.A transforming KIF5B and REF gene fusion in lung adenocarcinoma revealed from whole-genome and transcriptome sequencing[J].Genome Research,2012,22(3):436 -445.
[7] LANGMEAD B,SALZBERG S.Fast gapped-read alignment with Bowtie 2[J].Nature Methods,2012,9:357 -359.
[8] KAI W,MINGYAO L,HAKON H,et al.ANNOVAR:functional annotation ofgenetic variantsfrom highthroughput sequencing data[J].Nucl.Acids Res.,2010,38(16):e164.
[9] EMANUELA S,CLAUDIA B,Beatrice P,et al.A somatic mutation in the 5'UTR of BRCA1 gene in sporadic breastcancercauses down-modulation oftranslation efficiency[J].Oncogene,2012,s:4596 -4600.
