基于pose共享的蛋白质-配体构象并行搜索算法
2014-11-14杨伟,吕强
杨 伟,吕 强
(1.苏州大学计算机科学与技术学院,江苏 苏州215006;2.苏州大学江苏省计算机信息处理技术重点实验室,江苏苏州215006)
随着X射线衍射以及NMR等技术的发展,越来越多的蛋白质的三维结构被测定出来,使基于受体结构的计算机辅助药物设计更具现实意义。分子对接(Molecular docking)方法是基于受体结构的药物设计(Structure-based drug design,SBDD)中的重要方法之一。分子对接是指两个或多个分子通过几何匹配和能量匹配相互识别的过程,通过分子对接可以从已有药物分子库筛选出有希望的先导化合物,避免了繁琐的化学合成实验过程,缩短了药物研发周期,降低了研发成本[1]。分子对接还被用于重新评估已知药物,发现已知药物新的适应症[2]。近年来,科研人员把注意力转向天然药物,希望从中药中开发出更为安全有效的新药,中药成分复杂,分子对接成为科学阐述中药药效物质基础和作用机理成为中药研究的重要工具[3]。分子对接操作就是寻找配体与受体结合在受体活性位点处的低能构象的过程。对接算法的性能依赖于两个因素:能量函数的精度和复合物构象搜索算法的性能。分子对接所涉及的搜索空间非常巨大,即便对柔性小分子,粗略估计其搜索空间至少含1030个解,要从中找出低能构象必须借助于各种优化算法。目前,已经有许多优化算法用来解决分子对接问题,典型的模拟退火算法、遗传算法、禁忌搜索算法、蒙特卡罗方法以及这些算法的各种修正变种[4-6]。
常用的分子对接模拟软件有 AUTODOCK[10],RosettaLigand[7],GLIDE[12], DOCK[13],FIexX[14],GOLD[15]等。Rosetta是华盛顿大学开发的开源生物大分子建模软件包,其中包含用于对蛋白质和核酸进行结构预测、设计和重建模的工具,在Rosetta中,分子的结构用pose数据结构表示[4]。RosettaLigand是其中利用模拟退火算法进行蛋白质-配体柔性对接的程序工具。本文的研究基于Rosetta v3.4版,下文中提及RosettaLigand处均指Rosetta v3.4中的RosettaLigand对接程序。Rosetta源代码可通过http://www.rosettacommons.org/网站获取。
1 材料与方法
RosettaLigand的对接算法流程如图1所示[6]。

图1 RosettaLigand对接算法Fig.1 Original dock algorithm in RosettaLigand
在Rosetta框架内,RosettaLigand对接协议被重复启动N次(串行或并行),生成指定数量的侯选结构,称为decoys。在这次N次轨迹中,RosettaLigand对接实例之间并不共享采样信息,彼此之间完全独立。我们认为在多次对接实例之间共享pose信息可以帮助RosettaLigand更好的对接蛋白质与配体。RosettaLigand搜索蛋白质-配体复合物构象的过程本质是在一个在Rosetta全原子能量函数的指导下,在能量地形图上搜索的过程。由于蛋白质-配体复合物自由度非常大,而且已知能量函数并不足够精确,所以不存在全局最优结构的精确表达式,通常对接实验都会生成数目巨大的侯选结构,以期能够采样到尽可能多的近天然结构,然后通过基于能量若者机器学习的挑选方法将近天然结构挑选出来[9]。所以对接程序的搜索构象的能力依赖两个因素,一是搜索的广度,二是搜索的深度和充分性。RosettaLigand通过运行多次对接轨迹来解决搜索的广度问题,我们现在希望通过在多个轨迹之间共享采样信息来改善RosettaLigand的构象采样的深度和充分性问题,如图2a,每一个点代表一次对接过程,在不共享采样信息的情况下,每个对接实例只能在其附近空间进行不充分的构象搜索,而在图2b中,大量的搜索实例某个进行吸引到构象空间最能较低处,并对此处进行充分的采样。
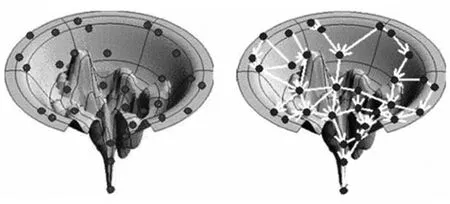
图2 共享与非共享采样信息搜索示意图Fig.2 Searching pattern:sampling sharing vs non-sharing
RosettaLigand在对接的DockMCM阶段使用MonteCarlo判断接受或拒绝当前采样。在经过修改的RosettaLigand算法中使用MonteCarlo判断接受或拒绝采样时,首先将其他对接实例以文件形式共享的最佳采样结果读入,与当前采样结果进行比较,而不是与RosttaLigand内置算法中实现的与本实例已采样的最佳结果比较。经过修改的DockCMC算法流程如图3所示:
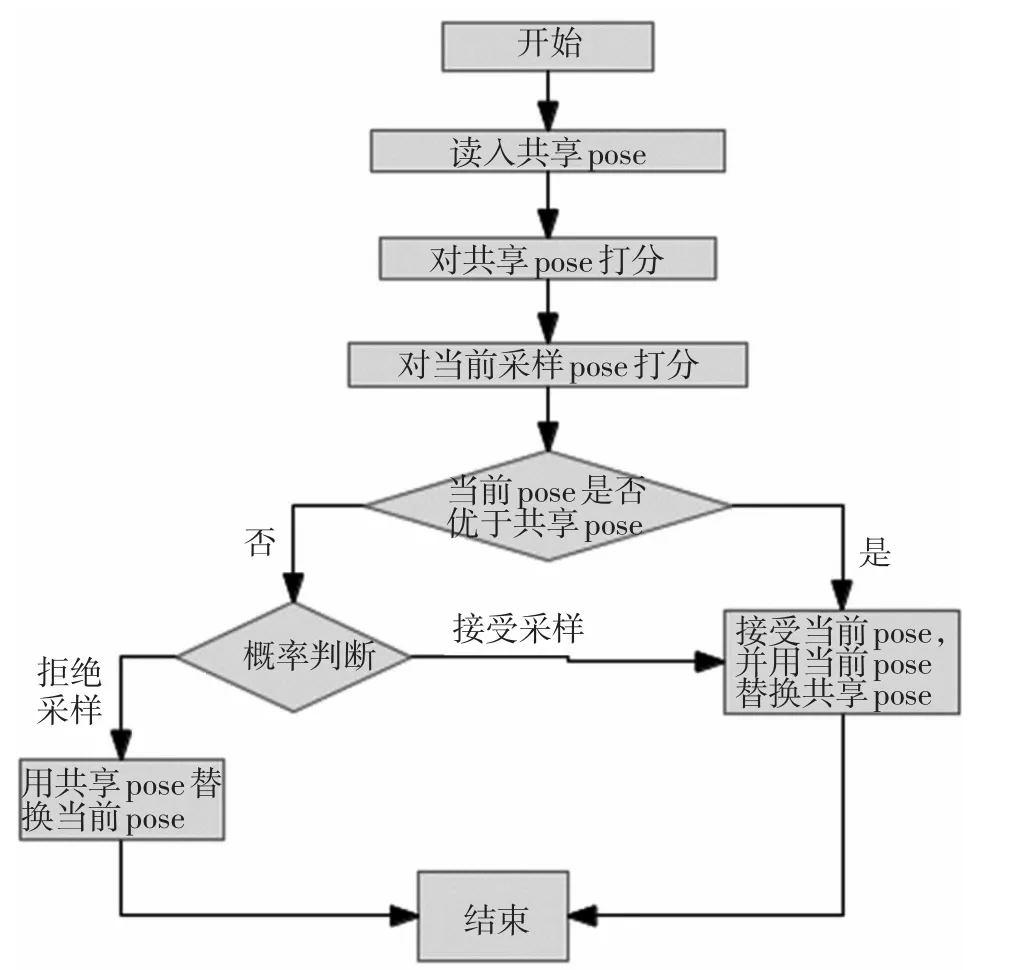
图3 RosettaLigand DockCMC的改进流程Fig.3 Modification to original RosettaLigand DockMCM
RosettaLigand原始对接算法在生成一个候选结构的采样过程中保有一个last_accepted_pose对象,用于存储上一次模拟退火接受的采样结果。在每一次新的采样后,将当前采样结果与此last_accepted_pose比较:
如果当前采样结果优于last_accepted_pose,则接受此次采样,并用此采样结果替换last_accepted_pose对象;如果如果当前采样结果不如last_accepted_pose,则按概率接受或拒绝当前采样结果,接受时同样用当前采样结果替换last_accepted_pose,拒绝时用last_accepted_pose做为下次采样的起点。
在共享pose的RosettaLigand对接算法中,同样会保有一个 last_accepted_pose对象,与原始的RosettaLigand算法不同,在每一次新的采样结束后,采样结果不是与上一次接受的采样结果比较,而是与共享的采样结果进行比较:如果当前采样被接受,则用当前采样结果替换共享的采样结果;如果当前采样被拒绝,则用共享的采样结果做为下一次采样的起点。
修改过的采样接受判定算法如下:

本文从meiler数据集[8]中选择11个自对接的算例进行实验,这11个目标是1AQ1,1DBJ,1DM2,1NJA,1NJE,1PB9,1PBQ,1Y1M,2AYR,2PRG 和4TIM。
配体的异构体库使用OMEGA2[11]生成,实验组为16个并行进程以共享采样信息方式生成5 000个侯选结构,对照组使用Rosetta平台的内置的MPI框架使用16个进程生成5 000个侯选结构。计算平台为16核SMP Linux集群。
2 实验结果
图4所示为11个目标的候选结构中Ligand-RMSD落在0~1和1~2两个区间的累积直方图。从图中可以看出,1AQ1、1DBJ、1PBQ、1Y1M、2AYR这5个目标,共享pose算法极大的提高了侯选结构集合中 Ligand-RMSD小于 2.0的比例(Ligand-RMSD小于2.0的构象被认为是近天然构象);1NJA和1NJE这两个目标,共享 pose算法和RosettaLigand内置算法表现都很差;而 1DM2、1PB9、2PRG、4TIM这4个目标,虽然共享pose算法不如RosettaLigand算法,但性能相近。表1统计了11个目标的候选结构中Ligand-RMSD小于2.0的构象的数量、构象最低能量以及实验运行时间。从表中可以看出,除了目标1Y1M,对于其他所有目标,共享pose算法都采样到了比对照组更低的能量,而计算时间平均仅增加了不到10%。

表1 对照组与实验组候选结构集合中Ligand-RMSD小于2.0的数量、最低能量值以及运行时间统计Table 1 Comparison of number of decoys with lrmsd <2.0,lowest energy and duration between experimental and control group
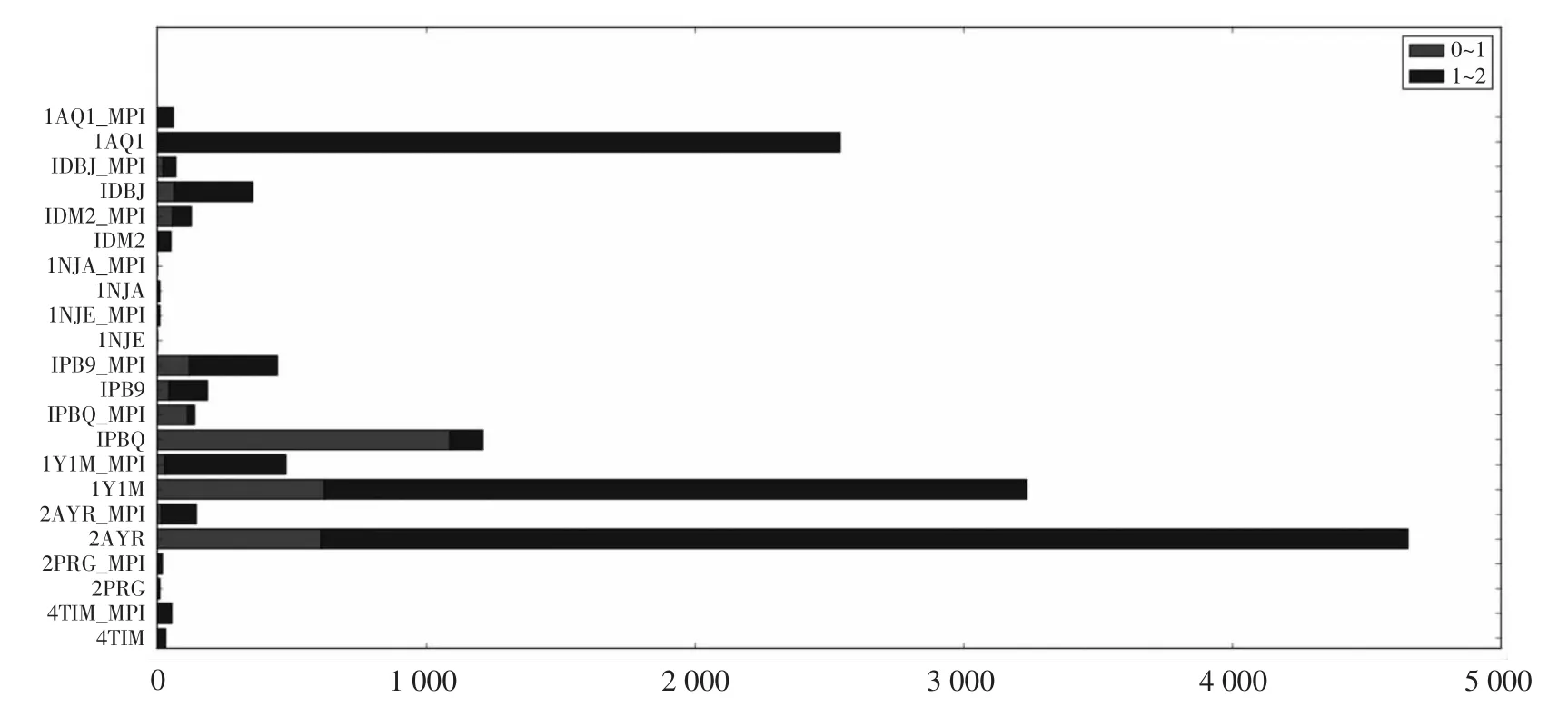
图4 候选结构集合中Ligand-RMSD落在0~1和1~2两个区间的累积直方图Fig.4 Stacked histogram of decoys with lrmsd fallen in interval 0~1 and 1~2

图5 11个目标候选结构总体能量值分布直方图Fig.5 Histogram of Rosetta energy of all decoys of all 11 targets
图5所示为11个目标的5 000个侯选结构的能量分布的直方图。从图中可以看出,除1Y1M外,对大多数目标来说,与RosettaLigand内置算法相比,共享pose算法使降低了侯选结构整体的平均能量。
3 结论
RosettaLigand以多次随机启动方式来解决构象搜索的广度问题,此方式类似于在崎岖的能量地形图上随机分布起始点,并在每一个起始点附近使用模拟退火进行构象搜索,搜索的性能依赖随机启动的次数与随机程度。我们的方法在N个并行的对接实例之间共享采样信息,当某个进程采样到一个近天然构象时,其他的对接实例会被吸引到全局最优点附近,并对该处进行充分采样,这就是在目标1AQ1、1DBJ、1PBQ、1Y1M、2AYR 中看到的情况,近天然构象的数量远远超过对照组。并且,由于每个对接实例在每一次采样后都与会N个实例当前所采样到的能量最低的构象进行比较,所以绝大多数实验组生成的候选结构集合的平均能量低于对照组的平均能量。当然,我们也看到,目标1DM2、1PB9、2PRG、4TIM的实验组采样到的近天然构象数量低于对照组,分析其原因是共享信息一方面强化了并行对接实例在能量地形图上某处的采样能力,但它同时另一方面也使得陷入局部最小的问题更加严重,原本在独立采样的情况下可能采样到近天然结构的实例可能被误导到非近天然区域。共享采样信息是有用的,但要避免陷入局部最小。如同样启动N个并行对接实例,在每个对接实例采样都收敛之后,输出采样结果,再比较当前N个实例采样所得结果,以能量最低的结果当作N个并行对接实例下一轮对接的起始结构,以此循环。这样在保证搜索广度的同时,又可兼顾搜索的深度。更进一步的,在共享整体pose的前提下,搜索的广度不可能超出非共享条件下搜索的广度,原因是无论如何选择共享的时机与策略,所共享的采样信息都是当前N个并行对接实例可能采样到的构象。所以,为了拓宽搜索的广度,以共享采样信息为基础,结合并行对接实例自身的采样结果来构造超出当前所有对接实例本次对接轨迹可能覆盖的构象空间的构象是一条可行的道路。
References)
[1] KAR S,ROY K.How far can virtual screening take us in drug discovery[J].Expert Opinion on Drug Discovery,2013,8(3):245-261.
[2] MA D L,CHAN D S H,LEUNG C H.Drug repositioning by structure-based virtual screening[J].Chemical Society Reviews,2013,42(5):2130-2141.
[3] 任洁,魏静.分子对接技术在中药研究中的应用[J].中国中医药信息杂志 ,2014,(1):123-125.REN Jie,WEI Jing.Application of molecular docking techniques in Chinese herbal medicine[J].Chinese Journal of Information on Traditional Chinese Medicine,2014,(1):123-125.
[4] KAUFMANN K W,LEMMON G H,DELUCA S L,et al.Practically useful:What the Rosetta protein modeling suite can do for you[J].Biochemistry,2006,49:2987-2998.
[5] 刘敏,曾涛,徐开阔,等.一种基于免疫遗传算法的分子对接构象搜索策略[J].计算机研究与发展,2009,46(z2):597-601.LIU Min,ZENG Tao,XU Kaikuo,et al.A molecular docking's conformational search strategy based on immune genetic algorithm[J].Journal of Computer Research and Development,2009,46(z2):597-601.
[6] 李纯莲,王希诚,赵金城,等.药物分子对接中的构象搜索策略[J].计算机与应用化学,2004,21(2):201-205.LI Chunlian,WANG Xicheng,ZHAO Jincheng,et al.Drug molecular docking design based on optimal conformation search [J].Computers and Applied Chemistry,2004,21(2):201-205.
[7] DAVIS I W,BAKER D.RosettaLigand docking with full ligand and receptor flexibility[J].Journal of molecular biology,2009,385:381-392.
[8] MEILER J,Baker D.ROSETTALIGAND:protein-small molecule docking with full side-chain flexibility[J].Proteins,2006,65,538-584.
[9] 杨凌云,吕强.一种基于SVR的分辨近天然GPCR―配体构象的方法[J].生物信息学,2011,9(2):167-170.YANG Lingyu,LÜ Qiang.A SVR-Based method for indentifying near-native GPCR-Ligand confromation decoys[J].China Journal of Bioinformatics,2011,09(2):167-170.
[10] MORRIS G M,GOODSELL D S,HALLIDAY D S,et al.Automated docking using a lamarckian genetic algorithm and and empirical binding free energy function[J].J Comp Chem,1998,19:1639-1662.
[11] HAWKINS P C,SKILLMAN A G,WARREN G L,et al.Conformer generation with OMEGA:Algorithm and validation using high quality structures from the protein databank and cambridge structural database[J].Journal of Chemical Information and Modeling,2010,50(4):572-584.
[12] FRIESNER R A,MURPHY R B,REPASKY M P,et al.Extra precision glide:Docking and scoring incorporating a modelofhydrophobic enclosure for protein-Ligand complexes[J].J.Med.Chem.,2006,49,6177-6196.
[13] LANG P T,BROZELL S R,MUKHERJEE S,et al.DOCK 6:combining techniques to model RNA-small molecule complexes[J].RNA ,2009,15(6):1219-1230.
[14] RAREY M,KRAMER B,LENGAUER T,et al.A fast flexible docking method using an incremental construction algorithm[J].J.Mol.Biol,1996,261:470-489.
[15] JONES G,WILLETT P,GLEN R C,et al.Development and validation of a genetic algorithm for flexible docking[J].J.Mol.Biol.,1997,267:727-748.
