LiH 的热力学性质及分子内部运动对体系热力学性质的影响
2014-07-13任维义
黎 波,任维义
(西华师范大学理论物理研究所,南充637002)
式 (3)中,M 为每个原胞中分子的质量,Bs 绝热体弹模量,Bs 可以近似写为[13]:
1 引 言
固态氢化锂(LiH)是电子结构最简单的离子晶体,它的晶胞中仅含四个电子,因此,它一直被当作检验固体量子理论中各种计算方法的理想体系.此外,氢化锂 (LiH)具有高熔点 (688℃),低密度(0.774g/cm3),高含氢量的特点,在真空和惰性气氛中,热力学相对稳定、不易分解.这些性质使氢化锂及其同位素化合物成为热核反应的重要原料和优异的储氢材料.因此,氢化锂及其同位素成为人们长期持续关注的课题之一.姜礼华等人[1]计算了LiH 在高压下的状态方程,鲁光辉等人[2]研究了LiH 的基态结构和势能函数,BW James等人[3]计算LiH 在低温下的弹性性质,Brodsky M H 等人[4]和Plekhanov V G 等人[5]从理论上计算了LiH 的晶格参数和电子结构,Yu Wen等人[6]研究 了LiH 从B1 相 (NaCl结 构)到B2 相(CsCl结构)的结构相变,并预测其相变压力为308GPa,Pandey J D 等人[7]计算了LiH 在不同温度下的宏观性质,如力常数和体弹模量等性质.然而对其在高温高压下热力学性质的研究还不是很充分,尤其是LiH 分子内部运动对体系的热力学性质的影响,还没有具体的报道.
本文采用基于密度泛函理论的第一性原理平面波超软赝势法 (UPS),结合准谐德拜模型,计算了LiH 在高温高压下的热力学性质,然后采用统计热力学的方法讨论了LiH 分子内部运动对体系热力学性质的影响 (以热容为例),并对其机理进行分析.
2 理论及计算方法
2.1 LiH 热力学性质的第一性原理计算方法
LiH 晶体是NaCl结构,属于Fm3m 空间群(No.225),具有中心原子,晶格常数a=b=c=0.4075nm,α=β=γ=90o[8],Li原子位于 (0,0,0)处,H 原子位于 (0.5,0.5,0.5)处,选取一个原胞进行计算.
在结构计算中,本文运用Castep程序[9],采用总能量平面波超软赝势方法[10],将离子势用赝势代替,电子波函数用平面波基组展开,用广义梯度近似(GGA)处理电子-电子间的交换相关势,选取Li和H 的价电子组态分别为2S1和1S1.在倒易K空间内,平面波截断能取300eV,计算精度选为fine,Monhorst-Pack网格取为6×6×6,交换相关函数取PBE函数[11],结构优化采用BFGS算法[12].为保证计算精度,每个原子的能量差小于10-5eV atom-1、原子间相互作用力低于0.03eV Å-1、原子公差偏移小于0.001Å 和应力收敛标准小于0.05GPa时则认为自洽收敛.
LiH 热力学性质中,我们采用了准谐德拜模型,即LiH 非平衡吉布斯函数G*(V;P,T)可以写为以下形式[13]:

在式 (1)右侧,E (V)表示LiH 每个原胞体积下的总能量,P,V 和T 分别为压强、体积和温度,Θ (V)为德拜温度,Avib为振动的Helmholtz自由能.考虑到准谐近似[14]和使用声子态密度的德拜模型Avib可以用下面等式表示为[15-17]:
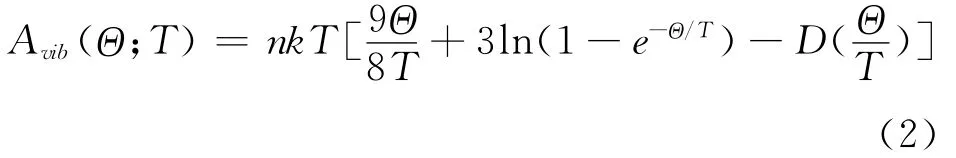
其中n为每个原胞中包含的原子数目,D (Θ/D)是德拜积分,k 为波尔兹曼常数.对于各向同性固体,德拜温度Θ 可以表示为[15]:
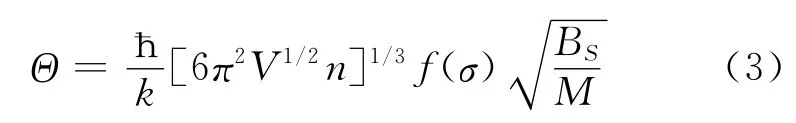
式 (3)中,M 为每个原胞中分子的质量,Bs绝热体弹模量,Bs可以近似写为[13]:

f(σ)由文献[16,17]给出;σ为泊松比 (Poisson Ration),取为0.25[18].因此,非平衡吉布斯函数G*(V;P,T)可以通过对体积求最小值简化为关于V 的函数,即:
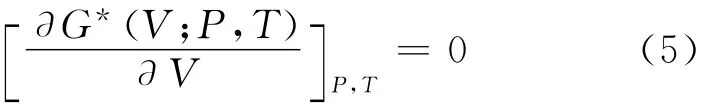
通过求解方程 (5),可以得到状态方程 (Equation of State,EOS)V (P,T),且等温弹性模量BT和热容CV,以及熵S和热膨胀系数α分别可以表示为:
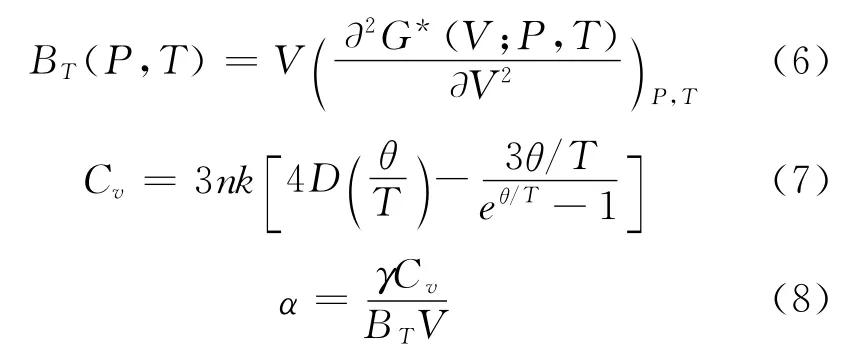
其中γ为Grüneisen参数,定义为:
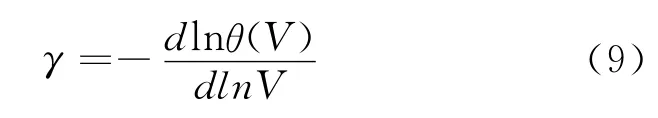
2.2 LiH 分子内部运动热力学性质的统计计算
分子的内部运动包括振动、转动、电子运动及核运动,LiH 体系是由大量LiH 分子组成的,因此,分子的内部运动对体系的宏观热力学性质都会产生贡献.平衡态唯象热力学的理论表明,热力学特性函数包括了平衡性质的全部信息.在统计力学中,配分函数起着特性函数的作用,一切热力学函数都可由配分函数求算[19].
根据配分函数的分解定理,LiH 分子内部运动配分函数qin可以写为[19]:

其中,qe为电子配分函数,qn为核配分函数,qv为振动配分函数,qr为转动配分函数.由于绝大多数双原子分子的基态光谱项是单重态,并且基态与第一激发态间的能差较大,在一般温度下,电子处于基态,故电子配分函数qe对分子内部运动配分函数无贡献,可视为一.实际上,核能级间距非常之大,其数量级至少是T=1010K 时的kT 值,这就是说,在通常温度下,核处在基态,则qn可表示为:
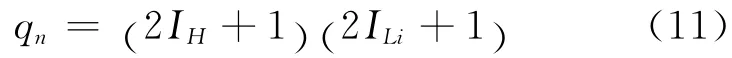
IH、ILi分别为H 原子和Li原子的核自旋量子数,值分别为0.5、1.5.
双原子分子转动和振动并非独立的,必须考虑其耦合,振-转配分函数为:

式 (12)中,δ 为对称数,LiH 为异核双原子分子,δ为1.v、J为振动和转动量子数,εv为双原子分子振动能量、εJ为双原子分子转动能量、εv,J为双原子分子振-转能量.孙卫国等人提出的双原子分子振-转能量表达式为[20]:
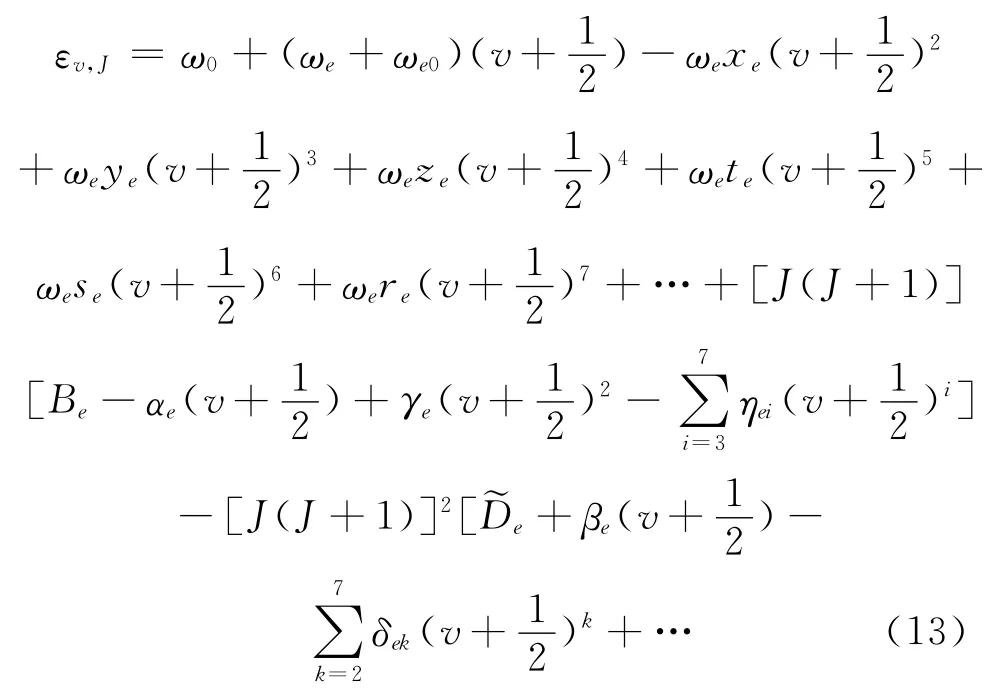
其中,LiH 分子的最大振动量子数vmax、振动光谱常数ω0、ωe、ωe0、ωexe、ωeye、ωeze、ωete、ωese、ωere和转动光谱常数Be、αe、γe、ηei 、~De、βe、δek由孙卫国等人[20,21]提出的代数方法(AM 方法)和势能变分法(PVM 方法)算得列于表1中.

表1 LiH 分子的振动光谱常数 (cm-1)、转动光谱常数 (cm-1)及最大振动量子数vmaxTable 1 The vibrational spectrum constants(cm-1),rotational spectrum constants(cm-1)and maximum vibrational quantum number vmaxof LiH molecules
由于高阶振动常数和转动常数很小,当转动能级不是很高时,可以将他们忽略,再将能量零点取在无转动的振动基态能级上,(13)式可以化简为以下两式:
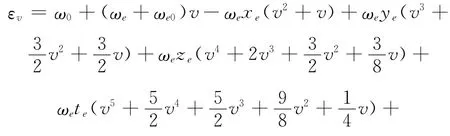
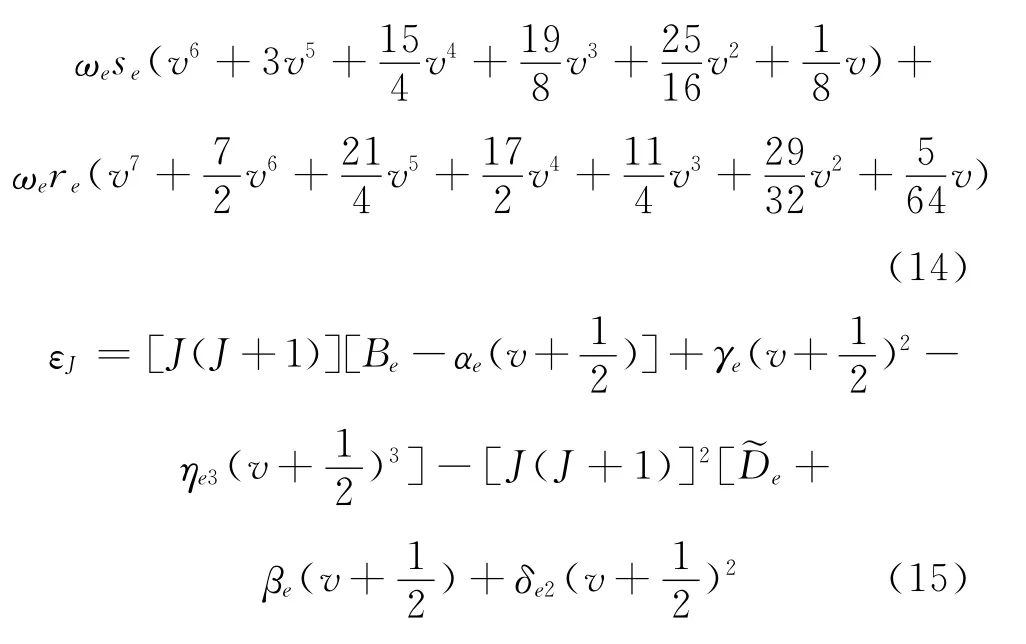

则 (15)式又可以化简为:
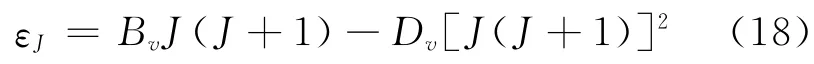
实际计算时,可以将转动求和部分qr(v)用Euler-Maclaurin公式做渐进展开为:

结合 (12)、(14)、(19)式得到双原子分子的振-转配分函数为:
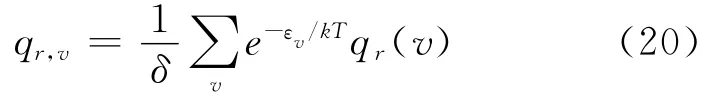
结合 (10)、(11)、(20)式就可以得到LiH分子内部运动的配分函数:

最后,由统计物理方法就可以得到LiH 分子内部运动对体系热力学性质的贡献:
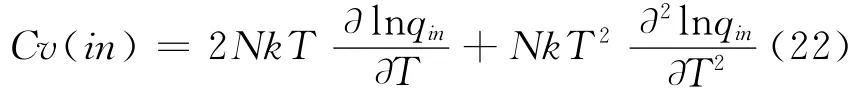
3 结果和讨论
3.1 LiH 的热力学性质
首先,分别设置一系列大小不同晶格常数的原胞模型,并计算与之相对应体系的总能量E 和原胞体积V,得到E 与V 的变化关系,再利用Birch-Murnaghan equation of state(EOS)[22]状态方程进行拟合,获得了平衡状态下的晶格常数和体弹模量,然后采用Gibbs程序[13]得到其相关的热力学性质.
图1给出了LiH 原胞总能量E与体积V 的变化关系图,从图1可以看出,在LiH 的结构模型中,在零温零压下,晶体原胞体系的能量达到最小值,此时V 为110.22Bhor3,说明系统存在稳定结构.表2给出了LiH 在零温零压下的晶格常数和体弹模量,其中晶格常数的计算值与实验值的误差仅为1.15%,且晶格常数和体弹模量与其他计算值符合得较好,说明这种计算方法是可靠的.
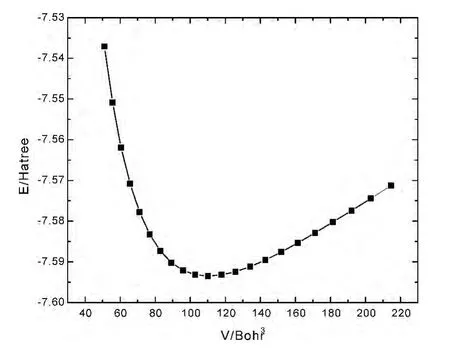
图1 LiH 的总能量E与原胞体积V 的关系Fig.1 Total energy E as a function of primitive cell volume V
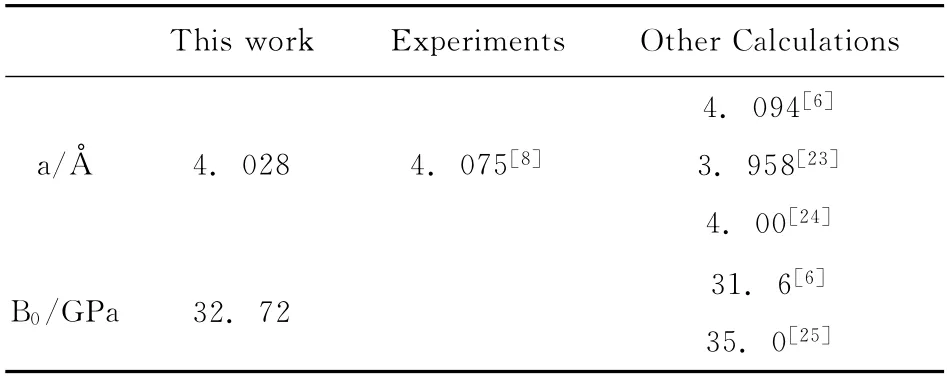
表2 LiH 在零温零压下的晶格常数 (Å)和体弹模量 (GPa)Table 2 Lattice constants(Å)and bulk modulus(GPa)of LiH at P=0GPa and T=0K
材料的热力学性质主要是材料在一定的温度和压力下,材料所表现出来的体积、热膨胀系数等物理量随温度和压强的变化及其变化规律.图2是LiH 在不同温度下,体积膨胀率 (V-V0)/V0随压强P的变化关系,图3是LiH 在不同压强下,体积膨胀率 (V-V0)/V0随温度T 的变化关系,图中正值表示晶体结构膨胀,负值表示晶体结构被压缩.从图2和图3中可以看出,在一定温度下,体积膨胀率随着压强的增加而减少.在压强一定时,体积膨胀率随温度的增加而增加.这说明对材料增加压强等同于对其降低温度,因为二者对材料引起的效应变化趋势是相同的.图2中,在压强不变时,温度越高,体积膨胀率的变化量越大,说明材料处在高温时,热运动更为剧烈,从而导致材料更容易被压缩.图3 中,在压强较低时,体积膨胀率随温度的变化明显,随着压强的增加,体积膨胀率随温度的变化非常缓慢,材料在压强的抑制作用下,增加温度很难使其膨胀,说明压强对晶体结构的影响比温度显著得多.
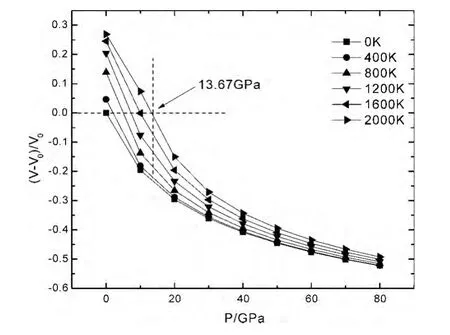
图2 LiH 在不同温度下体积膨胀率 (V-V0)/V0 随压强P的变化关系,其中V 表示任意压强下的体积,V0 表示零压下的体积Fig.2 Relationships between the volume expansion ratios(V-V0)/V0 and pressures P of LiH under different temperatures in which V is the volume under any pressure,V0indicates the volume under zero pressure
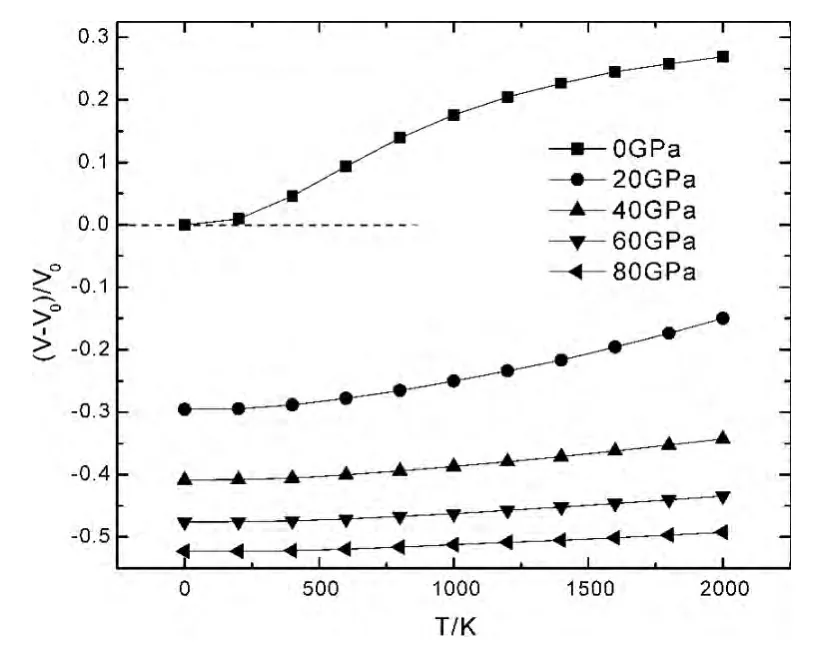
图3 LiH 在不同压强下体积膨胀率 (V-V0)/V0 随温度T 的变化关系Fig.3 Relationships between the volume expansion ratios(V-V0)/V0and temperatures T of LiH under different pressures
图4为LiH 在不同压强下,热膨胀系数α随温度T 的变化关系.在低温时,热膨胀系数随温度的增加迅速增大,在高温时,这种变化较为缓慢,当压强逐渐增加时,热膨胀系数随温度的升高的相对增加量逐渐减小,温度越高这种现象越明显;在给定温度下,低压时热膨胀系数随压强减小非常迅速,高压时热膨胀系数随压强的变化非常缓慢.图5为LiH 在不同压强下,德拜温度Θ 随温度T 的变化关系.从图中可以看出,德拜温度随着温度的增加而减小,变化趋势较为缓慢,但随着压强的增加而增大;300K 零压下的德拜温度为724.85K.

图4 LiH 在不同压强下热膨胀率ɑ随温度T的变化关系Fig.4 Relationships between the thermal expension coefficientsɑand temperatures T of LiH under different pressures
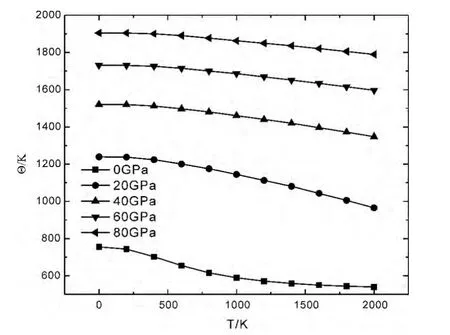
图5 LiH 得拜温度Θ 随温度T 的变化关系Fig.5 Relationships between the Debye temperaturesΘ and temperatures T of LiH under different pressures
最后,图6展示了LiH 的热容CV随温度T的变化关系.一定压强下的热容随着温度的增加而增加,且温度较低时 (T<800K),由于非谐效应的影响,热容随温度的增加较快;随着温度的升高热容增加越来越慢;当温度一定时,热熔随着压强的增加而减小,说明增大压强就等于降低温度.然而,随着温度和压强的不断升高,材料的热容几乎不发生变化,其热容CV=49.65J mol-1K-1接近Dulong-Petite极限值,即6NAk(≈49.90Jmol-1K-1).
3.2 分子内部运动对体系热力学性质的影响
利用LiH 分子的振动光谱常数和转动光谱常数,考虑分子振动和转动的耦合,得到LiH 分子更加精确的振-转配分函数,加上核配分函数、电子配分函数构成分子内部运动配分函数qin,其随温度的变化关系如图7所示,分子内部运动配分函数随温度按指数规律增加,温度越高增加得越快,占据的能量状态越大,对体系热力学性质的影响也越大.
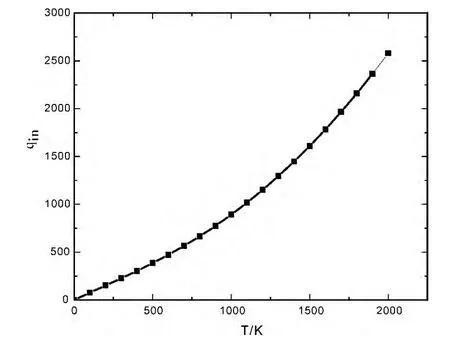
图7 LiH 分子内部运动配分函数qin 随温度T的变化关系Fig.7 The partition function of internal motion of LiH molecules qinas a function of temperature T
由LiH 分子内部运动配分函数,根据统计热力学理论计算得到了LiH 分子内部运动对体系热力学性质的贡献,如图8所示,Cv为0GPa时体系的热容量,CinV为分子内部运动对体系的贡献,CVCinV为体系势能对热熔的贡献.分子内部运动与压强无关,因此,用分子内部运动对热熔的贡献CinV与0GPa时体系的热熔值进行比较,分析分子内部运动对体系热力学性质的影响.从图中可以就看出,0-100K时分子内部运动对热熔的影响增加得较快,且占主要地位,体系势能对热熔几乎无贡献.100K-600K 范围内,分子内部运动对体系热熔的贡献缓慢增加,体系势能对热熔的贡献迅速增加,占主要作用.600K-2000K 时,体系热熔接近Dulong-Petite极限值,分子内部运动对体系热熔的贡献呈线性增加,较为缓慢,势能对体系热熔的贡献缓慢减小,可能是由于温度升高,电子和核跃迁到激发态,对热熔的贡献不能忽略,而本文忽略了电子和核激发态对热熔的贡献所致.

图8 LiH 分子内部运动对体系热力学性质的影响,其中Cv为LiH 在零压下的热熔,CinV 和CV -CinV 分别为LiH 分子内部运动及势能对体系热熔的贡献Fig.8 The influence of internal motion of LiH molecules on the thermodynamic properties of system,in which CinV is the heat capacity of LiH under zero pressure,CinV and CV-CinV are the effect of internal motion and potential of LiH molecules on the heat capacities of system,respectively
4 结 论
首先,利用基于第一性原理的平面波赝势密度泛函理论计算了LiH 在零温零压下的晶格常数和体弹模量,发现晶格常数和体弹模量与实验值及其他理论计算值符合得较好,当原胞体积为110.22Bhor3时能量最低,体系状态最稳定;然后,利用准谐德拜模型计算了LiH 在压强为0-80GPa,温度为0-2000K 范围内的体积膨胀率、热膨胀系数、德拜温度及定容热容.结果表明:体积膨胀率及热膨胀系数随着温度的增加而增大,随着压强的增大而减小,德拜温度随着温度的增加而减小,随着压强的增加而增加,对材料加压就等效于对其降温.在低温时热容随温度增加非常迅速,在高温时热容随温度的增加较为缓慢,热容随着压强的增加而减小,高温高压时热容趋近于Dulong-Petite极限值49.90Jmol-1K-1;最后,以孙卫国等人提出的代数方法 (AM)和势能变分法 (PVM),计算得到LiH 的振动能级和转动能级的完全集合为基础,采用统计热力学的方法计算了LiH 分子内部运动对体系热力学性质的影响.LiH 分子内部运动对体系热力学性质的影响是不可忽略的,以热容为例,随着温度的增加,分子内部运动对体系热容的贡献逐渐增加,在温度为0-100K 范围内,分子内部运动对热容的贡献占主要作用,在100K-2000K时,分子内部运动对体系热熔的贡献呈线性增加得趋势,增加得较为缓慢.
[1] Jing L H,Liu F S,Tian C L.Many-body interactions between ions in LiH crystal and its equation of state under high pressure[J].Acta Phys.Sin.,2008,57(7):4412(in Chinese)[姜礼华,刘福生,田春玲.LiH 晶体中离子间多体相互作用与高压下状态方程研究[J].物理学报,2008,57(7):4412]
[2] Lu G H,Sun W G,Fen H.Theoretic studies on the potential energy curves of some hydride diatomic molecules[J].Acta Phys.Sin.,2004,53(6):1753(in Chinese)[鲁光辉,孙卫国,冯灏.氢化物双原子分子势能曲线的能量自洽法研究[J].物理学报,2004,53(6):1753]
[3] James B W,Kheyrandish H.The low-temperature variation of the elastic constants of lithium hydride and lithium deuteride[J].J.Phys.C:Solid State Phys.,1982,15(31):6321.
[4] Brodsky M H,Burstein E.Infrared lattice vibrations of single crystal lithium hydride and of its isotopic derivations[J].J.Phys.Chem.Solids,1967,28:1655.
[5] Plekhanov V G.Isotope effects on the lattice dynamics of crystal[J].Mater.Sci.Eng.,2001,35:139.
[6] Yu W,Jin C Q,Kohlmeyer A.First principles calculation of phonon dispersion,thermodynamic properties and B1-to-B2phase transition of lighter alkali hydrides[J].J.Phys.:Condens.Matter,2007,19(8):6209.
[7] Pandey J D.Properties of isotopic lithium hydride in crystalline state[J].J.Inorg.Nucl.Chem.,1978,40:1184.
[8] Vidal J P,Vidal-valat G.Accurate debye-water factors of7LiH and7LiD by neutron diffraction at three temperatures[J].Acta Cryst.,1986,B42:131.
[9] Segall M D,Lindan P,Probet M J,et al.First-principles simulations:ideas,illustrations and the CASTEP code[J].Phys.Condens.Matter.,2002,14(11):2717.
[10] Vanderbilt D.Soft self-consistent pseudopotentials in ageneralized eigenvalue formalism[J].Phys.Rev.B,1990,41:7892.
[11] Perdew J,Burke K,Ernzerhof M.Generalized gradient approximation made simple[J].Phys.Rev.Lett.,1996,77:3865.
[12] Fischer T H,Almlof J.General methods for geometry and wave function optimization[J].Phys.Chem.,1992,96(24):9768.
[13] Blanco M A,Francisco E,Luana V.GIBBS:Isothermal-isobaric thermodynamics of solids from energy curves using aquasi-harmonic debye model[J].Comput.Phys.Commun.,2004,158:57.
[14] Maradudin A A,Montroll E W,Weiss G H,et al.Theory of Lattice Dynamics in the Harmonic Approximation[M].New York:Academic Press,1971.
[15] Blanco M A,Martín Pendás A,Francisco E,et al.Thermodynamical propertied of solids from microscopic theory:Applications to MgF2and Al2O3[J].J.Mol.Struct:Theochem.,1996,368:245.
[16] Francisco E,Recio J M,Blanco M A,et al.Quantum-mechanical study of thermodynamic and bonding properties of MgF2[J].J.Phys.Chem.,1998,102(9):1595.
[17] Florez M,Recio J M,Francisco E,et al.First-principles study of the rocksalt-cesium chloride relative phase stability in alkali halides[J].J.Phys.Chem.,2002,66(14):144112.
[18] Poirier J P.Introduction to the Physics of the Earth’s Interior[M].England:Cambridge University,1991.
[19] Gao Z D,Guo G L.Introduction to statistical thermodynamics[M].Beijing:Peking University Press,2004:134(in Chinese)[高执棣,郭国霖.统计热力学导论[M].北京:北京大学出版社,2004:134]
[20] Sun W G,Hou S L,Feng H,et al.Studies on the vibrational and rovibrational energies and vibrational force constants of diatomic molecular states using algebraic and variational methods[J].J.Mol.Spectrosc.,2002,215:93.
[21] Hou S L.Theoretical studies on vibrational and rotational spectra and dissociation energies of diatomic molecules[D].Chengdu:Sichuan University,2003(in Chinese)[候世林.双原子分子振转能谱和离解能的理论研究[D],成都:四川大学,2003]
[22] Murnaghan F D.The compressibility of media under extreme pressure[J].Proc.Natl.Acad.Sci.USA,1944,30:244
[23] Lebègue S,Alouani M,Arnaud B,et al.Pressureinduced simultaneous metal-insulator and structuralphase transitions in LiH:A quasiparticle study[J].Europhys.Lett.,2003,63(4):1.
[24] Hama J,Suito K,Kawakami N.First-principles calculation of the shock-wave equation of state of isotopic lithium hydrides[J].Phys.Rev.B,1989,39(5):3351.
[25] Roma G,Bertoni C M,Baroni S.The phonon spectra of LiH and LiD from density-functional perturbation theory[J].Solid State Commun.,1996,98(3):203.
