MgnLa(n=2-6)掺杂团簇的密度泛函研究
2014-07-13附青山余祖孝
附青山,余祖孝
(1.四川理工学院材料与化学工程学院,自贡643000;2.四川省腐蚀与防腐重点实验室,自贡643000)
1 引 言
近二十年来,原子团簇因其独特的性质被广泛的关注;多种的理论、实验方法被用来研究原子团簇的结构、电、光、磁等性质[1-7].团簇是由几个到几百个原子 (甚至几千个原子)所组成的凝聚体,是介于气态与凝固态 (液态、气态)之间的一种全新的过渡状态,具有许多特殊的性质,团簇的研究是介于原子分子物理和凝聚态物理之间的一个研究层次,其中有许多新的物理和化学现象日益引起人们的广泛关注.
对于镁团簇的研究已有较多的报道.Kumar等利用密度泛函理论研究了Mg团簇的稳定结构及键特性[6];Akola 等利用第一性原理结合密度泛函研究了小原子数Mg 团簇金属性的演生过程[8],指出小原子数Mg团簇由于s-p杂化以及导带与价带间能隙的缓慢演化使得其金属性的出现难以确定;Jellinek利用梯度修正的密度泛函理论研究了Mg原子团簇与Mg离子团簇的的稳定结构和电子特性,指出了团簇非金属性到金属性转变的尺寸依赖现象[9].然而,对于稀土元素掺杂的Mg团簇研究目前还较少.由于在Mg 中掺杂入稀土元素能使得Mg合金的性能得到较大的改善,使其合金在现代工业技术上具有较好的应用前景.因此,为了更好地从微观上探讨在Mg基中掺杂稀土元素的形成掺杂团簇的微观机理,我们选用La作为掺合原子,采用Gaussian03程序进行密度泛函理论 (Density Functional Theory,DFT)计算,研究了单La原子掺入Mg基中的二元金属团簇的结构和电子性质.为今后制备和检测团簇物质,以及对混合团簇的研究提供了最基础的理论参考.
2 计算方法
采用Gaussian03程序进行密度泛函理论DFT计算,用含有电子相关效应校正的DFT 中的B3LYP方法(由Becke建议的杂化交换函数和Lee Yang Parr相关函数组成),选择CEP-31g基组.自洽过程以体系的能量是否收敛为依据,能量收敛精度优于l0-6a.u..对MgnLa(n=2-6)的团簇,就各种不同的拓扑结构和可能的自旋多重度分别进行键长和键角的几何优化.为了节省时间和提高效率,优化分两个步骤:第一步给出一构形的键长、键角和二面角的初始值,定义较小的收敛精度(10-4a.u.)进行初步优化;再利用第一步优化出的值做初始值,将收敛精度提高到10-6a.u.再进一步进行结构和频率优化,能量最低,而且优化结果没有虚频的结构为平衡的基态结构.表1列出了MgnLa(n=2-6)团簇稳定几何结构的平均键长、对称性、原子化能、能级分布、能级间隙、束缚能、总能的二阶差分.
3 计算结果与讨论分析
3.1 团簇的几何结构
MgnLa(n=2-6)团簇的基态几何结构如图1所示,图中掺杂镧原子用浅色表示.图中显示出镧原子总是占据着团簇的表面位置,这可理解为是基于组成原子半径及相互之间键强关系,一价镧原子和三价镧原子的泡里电离半径分别为1.39a.u.和1.06a.u.,大于镁原子的泡里电离半径 (0.82a.u.),由于二原子系统的相关性,随着镁原子的增加,镧原子的低成键势使得镁原子更容易与其成键,并使镧原子趋于表面以改善体系的稳定性.
如图1所示,Mg2La的稳定构型是等腰三角形,镧原子占据三角形顶点的位置,镁-镁键长大于镧-镁键长,并由此开始了镧占据表面中心位置的构型.第一个三维结构出现在Mg3La,为四面三角锥体结构,镧占据在C3V构型的四面体顶点.经过频率的计算,Mg3La-B 和Mg3La-C 皆有虚频出现,为不稳定的过渡态结构,而Mg3La-A 为实频,说明四面体的结构是最稳定的构型.而Mg4小团簇的结构也是正四面体[10],表明镧的掺入将取代镁以前的晶体占位,而成为新的稳定构型.Mg4La-A 是Mg4La几种结构中能量最低的结构,它是由Mg5[10]的三角双锥构型镧取代一个镁原子而得来.从Mg3La-A 到Mg4La-A 只增加了一个镁原子,新增的镁原子首先与镧原子成键,而不是形成与镧相对的三角双锥,这表明镧原子具有更强的成键力,这是因为镧原子的电子结构 [Xe]5d16S2决定的.Mg5La的四种构型中,以Mg5La-A 的C4v四角双锥的构型能量最低,经过频率计算为实频的稳定态结构.而Mg5La-A 与Mg6[10]不同的是,在镧原子取代顶点的镁的同时,还能与四角双锥的另一个顶点的镁原子成键,由此进一步说明镧原子与镁原子之间的结合键力远强于镁与镁原子之间的键合力.而Mg5La-B 的构型虽然也是镧原子与5个镁原子成键,其结合能比Mg5La-A 构型更低,但是经过频率计算,Mg5La-B 为虚频,是不稳定的过渡态.
Mg6La的四种构型中,以Mg6La-A 的C5v五角双锥的构型能量最低,经过频率计算为实频的稳定态 结 构.而Mg6La-A 与Mg7[10]不 同 的是,在镧原子取代顶点镁原子同时,还能与五角双锥的另一个顶点的镁原子成键,镧-镁键长小于镁-镁键长,说明镧原子与镁原子之间的结合更稳定.而Mg6La-C、Mg6La-D 的构型虽然也是具有实频的稳定态结构,但结合能均高于Mg6La-A 的构型.
团簇的平均键长见表1,随镁原子数的增加,整体没有明显的单调趋势.在Mg3La和Mg5La处,分别出现了最小值和最大值.Mg3La是一个三棱锥构型,它在这系列的团簇中原子之间结合的更加紧密.
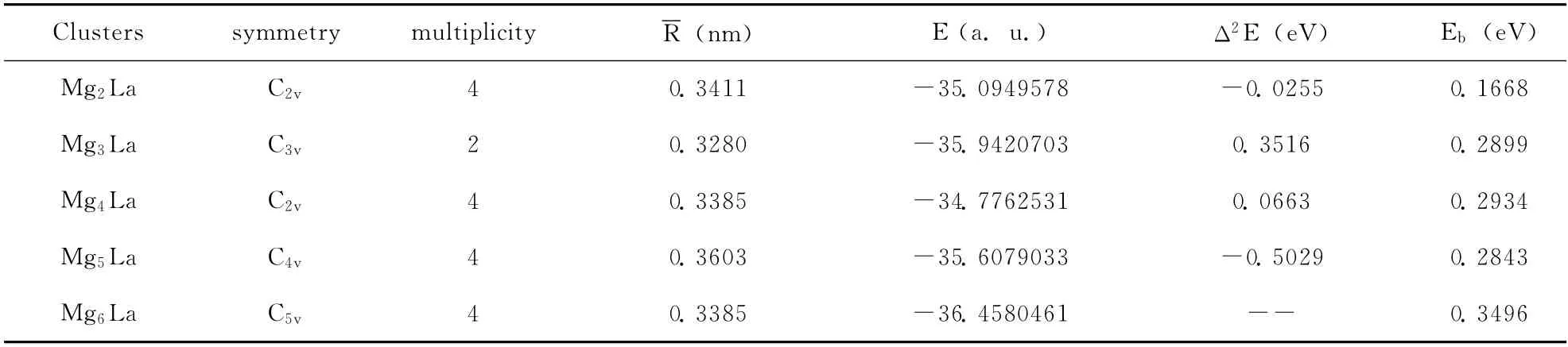
表1 MgnLa(n=2-6)团簇的几何和电子特性参数Table 1 The geometric and electronic characterisations of the MgnLa(n=2-6)clusters
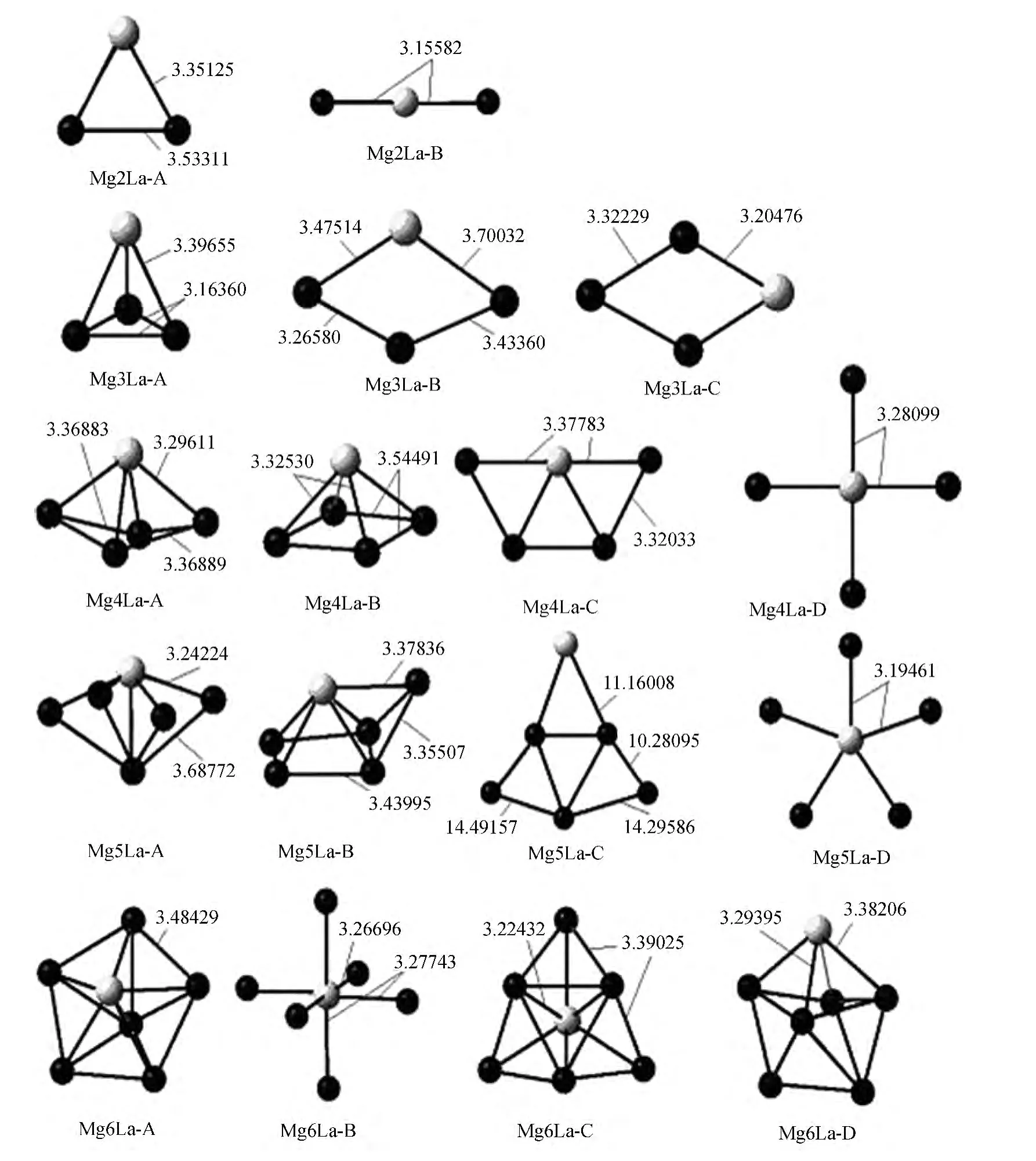
图1 MgnLa(n=2-6)团簇的构型,图中浅色为La原子,深色为Mg原子Fig.1 The geometric structures of MgnLa(n=2-6),La atoms are showed by the light colour and Mg atoms are showed by the deep colour
3.2 团簇的稳定性
团簇的稳定性可以用每个电子的束缚能Eb,能量的二阶差分Δ2E及来讨论,其表达式为:

众所周知,团簇的总能量的二阶差分Δ2E 是反映团簇稳定性的一个很敏感的量,其值越大,说明对应的团簇越稳定.MgnLa(n=2-6)团簇的Δ2E 随镁原子数的增加的变化如图2 (a)所示,从Mg2La到Mg3La快速增加,从Mg3La到Mg5La逐步降低,在Mg3La处有最大值,该结果说明Mg3La其稳定性比Mg2La,Mg4La,Mg5La团簇要高一些.Mg5La团簇的Δ2E 最低,说明它的稳定性在这几个团簇中最差.
团簇的每个原子的平均束缚能Eb反映团簇稳定性的另一个主要依据.如图2 (b)所示,随着镁原子数的增加总体是逐渐增大的,在Mg3La和Mg5La处出现拐点,Mg5La相比Mg3La和Mg4La稳定性有所降低.在Mg6La处有最大值,说明Mg6La的稳定性最高,其团簇的结构更稳定,但与块体的理论值还相差较远,对于表面效应突出的小团簇而言,其束缚能收敛于块体材料显然是缓慢的.综合以上的结果,与我们前期的工作对比[10],我们发现Mg6的最稳定构型是一个四棱双锥(D4h),而本文中Mg5La团簇稳定结构的对称性为C2v,表明La原子的掺入使得团簇的对称性降低,稳定性也减小.此外,对比Mgn(n=2-7)团簇,我们发现La原子的掺入主要是以取代的形式进行,占据团簇的表面位置.因此,La原子的掺入没有显着改变Mg团簇的稳定结构构型,只是由于Mg-Mg键与Mg-La键键长以及相互之间的不同作用造成稳定结构畸变,使对称性降低.从有限的结果可以看出Mg6La具有较高的稳定性,表明La掺杂的镁小团簇将以五角双锥的构型为主.

图2 团簇的 (a)总能量的二阶差分Δ2 E及 (b)每个原子的平均束缚能Eb随Mg原子数的变化规律Fig.2 (a)The second difference in energy and(b)the binding energy per atom of MgnLa(n=2-6)cluster as a function of the number of Mg atoms
3.3 团簇的能级分析
团簇的费米能级,如图3(a)所示,团簇的费米能级从Mg2La到Mg3La快速降低,在3个镁原子的团簇处最低,从Mg4La到Mg6La基本不变,这与团簇的壳层分布有关;Mg原子的价电子为2,La原子的价电子为3,Mg4La到Mg6La团簇的壳层轨道的电子排布分别为:1s21p62s21d1,1s21p62s21d3,1s21p62s21d5,从Mg4La到Mg6La增加的电子排布在1d轨道上,1d为5个简并轨道,因此Mg4La到Mg6La的费米能级基本不变.
团簇最高占据轨道 (HOMO)能量与最低空轨道(LUMO)能量的差值作为能隙 (Eg),团簇的能隙变化如图3 (b)所示,从Mg2La 到Mg5La团簇能隙逐步降低,在含5个镁原子的团簇处达到最低,然后又开始增加.Mg5La团簇具有较高的费米能级和较小的能隙,因此它具有较好的化学活性,而稳定性较差.这与前面团簇稳定性分析的结果是一致的.
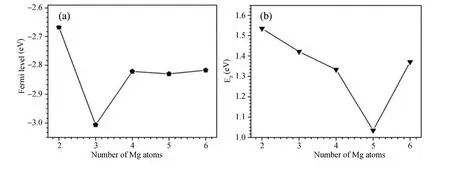
图3 团簇的 (a)费米能级及 (b)最高占据轨道和最低未占据轨道之间的能隙Eg随Mg原子数变化的规律Fig.3 (a)The fermi level and(b)the Eg(HOMO-LUMO gap)as a function of the number of Mg atoms in clusters
4 结 论
本文用密度泛函理论中的局域自旋密度近似方法研究了在Mg 基中掺入La形成掺杂小团簇MgnLa(n=2-6)的基态结构和电子性质.研究结果表明:掺入的La原子更易于趋于团簇的表面位置,且La原子的掺入多采用取代稳定Mg团簇上的Mg原子位置的方式.总能量的二阶差分、每个原子的平均束缚能、费米能级和能隙没有明显的单调趋势.在MgnLa(n=2-6)团簇中,Mg3La和Mg6La更加稳定;La掺杂的镁小团簇将以五角双锥的构型为主.
[1] Kakemam J,Peyghan A A.Electronic,energetic,and structural properties of C-and Si-doped Mg12O12nano-cages[J].Comput.Mater.Sci.,2013,79(0):352.
[2] Wang M,Huang X,Du Z,et al.Structural,electronic,and magnetic properties of a series of aluminum clusters doped with various transition metals[J].Chem.Phys.Lett.,2009,480(4–6):258.
[3] Chuang F C,Wang C Z,Ho K H.Structure of neutral aluminum clusters Aln(2≤n≤23):Genetic algorithm tight-binding calculations[J].Phys.Rev.B,2006,73(12):125431.
[4] Lyalin A,Solov’yov I A,Solov’yov A V,et al.Evolution of the electronic and ionic structure of Mg clusters with increase in cluster size [J].Phys.Rev.A,2003,67(6):063205.
[5] Acioli P H,Jellinek J.Electron Binding Energies of Anionic Magnesium Clusters and the Nonmetal-to-Metal Transition[J].Phys.Rev.Lett.,2002,89(21):213402.
[6] Kumar V,Car R.Structure,growth,and bonding nature of Mg clusters[J].Phys.Rev.B,1991,44(15):8245.
[7] Jia J,Wang J Z,Liu X,et al.Artificial nanocluster crystal:Lattice of identical Al clusters[J].Appl.Phys.Lett.,2002,80(17):3186.
[8] Akola J,Rytköen K,Manninen M.Metallic evolution of small magnesium clusters[J].Eur.Phys.J.D,2001,16(1):21.
[9] Jellinek J,Acioli P H.Magnesium Clusters:Structural and electronic properties and the size-Induced nonmetal-to-metal transition[J].J.Phys.Chem.A,2002,106(45):10919.
[10] Fu Q S,Sheng Y,Tu M J.Geometical and electronic properties of Mgn(n=2~7)clusters[J].J.Sichuan Univ.:Nat.Sci.Ed.,2006,43(05):1056(in Chinese)[附青山,盛勇,涂铭旌.Mgn(n=2~7)团簇的几何结构研究[J].四川大学学报(自然科学版),2006,43(05):1056]
