经由线粒体损伤诱发的药源性肝损伤研究进展
2014-03-09杨婷婷江振洲张陆勇
杨婷婷,江振洲,张陆勇
(1.中国药科大学江苏省新药筛选重点实验室, 江苏 南京 210009; 2.中国药科大学江苏省药效研究与评价服务中心, 江苏 南京 210009; 3.中国药科大学药物质量与安全预警教育部重点实验室, 江苏 南京 210009)
经由线粒体损伤诱发的药源性肝损伤研究进展
杨婷婷1,2,江振洲1,3*,张陆勇1,2
(1.中国药科大学江苏省新药筛选重点实验室, 江苏 南京 210009; 2.中国药科大学江苏省药效研究与评价服务中心, 江苏 南京 210009; 3.中国药科大学药物质量与安全预警教育部重点实验室, 江苏 南京 210009)
线粒体是物质氧化和能量转换的场所,在能量代谢及自由基的产生、衰老、凋亡中起着重要作用。线粒体的呼吸链缺陷、代谢酶失活、结构改变、基因突变等因素都会影响整个细胞的正常功能,从而导致病变的发生。线粒体是药物毒性作用的重要靶标,肝脏作为药物代谢的重要脏器,也是药物引发损伤的主要靶器官。一些抗病毒药物、抗肿瘤药物和抗生素等可显著诱导肝脏线粒体损伤。药物主要通过改变线粒体结构、酶的活性或减少mtDNA的合成,进一步破坏β-脂质氧化和肝细胞的氧化能力,最终诱发肝损伤。综述药源性肝损伤领域有关线粒体损伤的研究进展,为预防和诊断药源性肝损提供思路。
线粒体;线粒体损伤;药源性肝损伤;肝毒性;临床药物
药源性肝损伤(drug-induced liver injury, DILI),指暴露于药物而无其它感染性因素引起的肝功能损伤,是肝脏生化指标异常的常见原因之一。肝脏是作为外因性化学物质的药物进行代谢的重要脏器,更是药物引发损伤的主要靶器官,多数药物在肝脏各种酶的作用下转变为水溶性较强的物质从肾脏排出机体。
随着各种新药的不断研发和广泛应用以及临床联合用药的增多,药源性肝损伤的发生率呈现逐步增高的趋势。药源性肝损伤是造成药物研发失败、药物使用受限甚至撤离市场的主要原因之一,也是引发肝脏疾病的重要原因之一。药源性肝病约占药物不良反应的6%,是除心血管不良反应外,药物上市后被撤回的最主要原因[1]。有报道显示DILI的发生率为0.001%~0.01%,但由于DILI不但存在发现和诊断困难的情况,而且能观察到的暴露人群也远不齐全,故推测实际临床发生率远远高于报道的数量。同时,随着对应用中草药、植物药及保健品引起的肝损伤报道越来越多,而DILI的治疗措施目前仍主要限于立即停药、支持治疗和监测预防急性肝功能衰竭的发生,DILI的早期诊断与治疗存在着理论与技术上的瓶颈。因此,DILI对制药工业和药品监督机构是一个严峻的挑战[2]。
线粒体是普遍存在于哺乳动物成熟红细胞以外的真核细胞中的细胞器,是细胞进行氧化和能量转换的主要场所,是细胞的“动力工厂”。线粒体在哺乳动物的肝脏细胞中含量非常丰富,其参与肝细胞内电解质稳态平衡的调控、离子跨膜转运、氧自由基生成、细胞信号转导和细胞凋亡等一系列的细胞生理活动[3]。同时,线粒体也是最容易受损的一个细胞器,其可以显示细胞受损的程度。越来越多的研究表明,线粒体损伤可能是药源性肝损伤诱发因素和经由途径。因此,认识和深入研究线粒体损伤在药源性肝损伤发生中的重要作用,可为DILI临床早期诊断、预防及治疗提供新思路。
1 线粒体的结构与功能
1.1 线粒体结构
线粒体是细胞内除细胞核外另一个由双层膜构成的细胞器,其拥有一套独特的DNA分子和完整的遗传信息传递和表达系统。从外向内分别是外膜、膜间质、内膜和基质4个区室,与线粒体功能相关的上千种生物大分子都分别组装在不同的线粒体膜系结构和空间内。外膜是其最外层的单位膜结构,上面含有孔蛋白,此蛋白通道通过开关作用对进出线粒体的物质进行初步筛选;内膜除含有多种转运系统外,还含有大量的合成ATP的装置,细胞色素酶是其标志酶类;基质中的酶类最多,三羧酸循环、脂肪酸氧化、氨基酸降解等有关的酶都存在于基质中,此外,基质中还含有线粒体的遗传系统,包括DNA、RNA、核糖体和转录、翻译遗传信息所需要的各种装备。一般动物细胞线粒体的数目从数百到数千个不等,代谢旺盛的细胞中线粒体数目较多,如人的心肌、小肠、肝脏等细胞中线粒体含量很丰富,在肝脏细胞中约占胞质的20%,在心肌细胞中该比例超过50%,其在细胞内分布不均一,常集中在代谢活跃的区域部位。
1.2 线粒体的功能
线粒体是物质氧化和能量转换的场所,其主要功能是进行三羧酸循环及氧化磷酸化合成ATP,为细胞生命提供直接能量。此外,线粒体还与细胞中自由基的生成,调节细胞氧化还原电位和信号转导,调控细胞凋亡、基因表达、细胞内多种离子的跨膜转运及电解质稳态平衡,特别对细胞中Ca2+的稳态调节等有关。
2 线粒体损伤及其机制
2.1 线粒体损伤
线粒体损伤主要表现在4个方面:其一,线粒体呼吸酶活性的降低,解偶联剂、腺苷酸转位体引起的质子漏使得线粒体膜电位△Ψm降低,从而导致ATP合成的减少;其二,呼吸链电子传递减慢使活性氧(reactive oxygen species,ROS)产生绝对增多或抗氧化酶活性的降低使得ROS的相对量增多;其三,线粒体膜电位降低造成线粒体对Ca2+的摄取量减少或Ca2+的外流量增大,造成胞内Ca2+的紊乱,进一步影响Ca2+相关酶活性的调节和信号转导,如诱发内质网应激等;其四,经线粒体通透性转变孔(mitochondrial permeability transition pore, mPTP)开放或其他非依赖mPTP通路介导的细胞色素c(Cytc)的释放,激活caspase级联放大反应,最终诱导细胞的凋亡。
2.2 线粒体损伤的机制
2.2.1 氧化应激 氧化应激的本质是ROS增加和抗氧化剂缺失,导致体内氧化系统和抗氧化系统失衡。当超过机体抗氧化系统对ROS的清除能力时,体内过多的ROS会导致脂质过氧化、蛋白质变性以及诱发基因突变,最终导致细胞的氧化损伤。线粒体不仅是细胞内ROS产生的主要场所,也是ROS攻击的主要靶标。因此,当锰超氧化物歧化酶(manganese superoxide dismutase,Mn-SOD)、谷胱甘肽和谷胱甘肽过氧化物酶等线粒体抗氧化酶的抗氧化能力降低或线粒体ROS生成增多时,就会导致氧化应激[4]。氧化应激不仅抑制线粒体呼吸活性,减缓呼吸链的电子传递,进一步增加ROS的产生,还可以上调解偶联蛋白的表达[5],进一步引起质子漏使得线粒体膜电位降低,最终减少ATP的合成。
2.2.2 钙紊乱 正常生理条件下,线粒体对Ca2+的摄取和外排共同维持细胞内Ca2+的稳态,并与内质网、细胞膜共同参与调节胞浆内游离Ca2+的浓度,维持细胞内Ca2+的稳态。任何影响线粒体呼吸链复合物活性的因素、影响线粒体膜电位的因素、mPTP的开放、线粒体DNA突变等都可造成线粒体Ca2+的紊乱[4]。钙稳态失衡会对线粒体和细胞中黄素腺嘌呤二核苷酸-甘油磷酸脱氢酶、丙酮酸脱氢酶磷酸酶、烟酰胺腺嘌呤二核苷酸异柠檬酸脱氢酶等的多种酶活性造成影响[6]。胞浆Ca2+水平升高引起线粒体经由Ca2+单转运蛋白对Ca2+的摄取增加,抑制ATP合成,最终引起氧化磷酸化过程障碍。
2.2.3 线粒体生物合成减少 线粒体生物合成是生物组织中调控并决定线粒体成分与数量的一个动态又复杂的过程,涉及线粒体基因与核基因表达的协调,线粒体DNA的表达及核基因编码产物运输进入线粒体等。已知过氧化物酶体增殖物激活受体γ辅助活化因子1α(peroxisome proliferators-activated receptor gamma coactivator 1 alpha, PGC-1α)是线粒体生物合成的主要调节因子。它通过刺激核呼吸因子(nuclear respiratory factor 1, NRF-1)和线粒体转录因子A(mitochondrial transcription factor A, mtTFA)表达,使编码线粒体蛋白的基因表达上调,线粒体生物合成增加[7]。当PGC-1α基因转录水平和表达均明显降低时,线粒体合成减少,最终可诱发线粒体损伤。
2.2.4 线粒体通透性改变 线粒体通透性改变(mitochondrial permeability transition, mPT)是指线粒体内膜非特异性大孔道即通透性改变孔的非特异性开放,这是由于mPTP开放所致,mPTP是由位于线粒体膜上的多蛋白所形成的非选择性复合孔道,环孢素A能与其结合,阻止mPTP的开放。当mPTP呈病理状开放时,线粒体内膜外的H+大量回流至基质,内膜去极化而电位崩溃,氧化磷酸化解偶联,ATP合成停止;同时线粒体基质外流,大量超氧离子生成,基质渗透压升高而使线粒体肿胀,最终导致线粒体外膜破裂,释放Cytc和凋亡诱导因子等,通过激活caspase信号通路最终引起细胞死亡。
2.2.5 线粒体DNA突变 线粒体具有自己的遗传物质——线粒体DNA(mitochondrial DNA, mtDNA),但是由于mtDNA是裸露的,缺乏保护,且处于线粒体呼吸链产生的高活性氧的环境中,因此,mtDNA极易受氧自由基的侵害[8]。
3 药物经由线粒体诱发肝损伤的途径
药物诱导的线粒体损伤主要包括结构改变和功能紊乱。线粒体结构的改变主要表现为线粒体肿胀、体积显著增大、内外膜完整性破坏甚至出现膜溶解、线粒体脊断裂及大量空泡化,线粒体结构基本丧失。药物经由线粒体诱发肝损伤的机制主要有干扰线粒体呼吸链上的电子传递、氧化磷酸化的解偶联等5个途径(见图1)[9]。

图1 药物致线粒体损伤的可能靶点和途径Figure 1 The possible targets and pathways of druginduced mitochondrial damage
3.1 干扰线粒体呼吸链上的电子传递
电子传递链(electron transfer chain, ETC)是典型的多酶体系,是线粒体内膜上存在传递电子的一组酶的复合物,是由一系列能可逆地接受、释放电子或H+的化学物质所组成,它们在内膜上相互关联地有序排列成传递链,伴随着营养物质的氧化产能。电子传递链的抑制在某种程度上意味着细胞能量代谢障碍,甚至细胞生命的终结[10]。
Tai等[11]在体外实验中发现,抗凝血药物氯吡格雷在10 mg·L-1的剂量下能明显抑制肝细胞线粒体的Ⅲ态呼吸和Ⅳ态呼吸,并且能延长Ⅲ态呼吸的基础耗氧,明显下调线粒体ATP合成能力,作者认为10 mg·L-1高剂量下的氯吡格雷对氧化磷酸化的抑制在于其干扰了电子传递链,进而影响线粒体的呼吸功能。
Cytc是线粒体呼吸链中传递电子的载体。Cytc释放入胞浆说明了ETC的严重损伤,电子传递障碍导致ROS生成增加,最终引发细胞凋亡,而Cytc缺失或其功能障碍也会导致呼吸链电子传递中断,加剧细胞损伤[11]。Zahno等[12]在对氯吡格雷诱发肝损伤的研究中发现,氯吡格雷主要通过CYP3A4代谢,其对肝细胞的损伤与其代谢物有关,后者能诱导肝细胞内ROS含量增加,Cytc释放,同时出现线粒体的损伤,而谷胱甘肽能降低氯吡格雷对肝细胞的毒性。
对乙酰氨基酚是临床最常用的解热镇痛抗炎药,也是临床引起肝毒性的最常见的药物,长期或过量使用该药易导致急性肝功能衰竭。线粒体是对乙酰氨基酚诱导肝毒性的靶细胞器,是最早发生形态学和功能改变的细胞器之一,其对肝细胞线粒体的损伤也是多途径的:经发现在过量服用对乙酰氨基酚2~6 h出现线粒体膜电位崩溃,加重线粒体的损伤[13];Kaushal等[14]发现对乙酰氨基酚能显著降低肝细胞中Na+-K+-ATP酶的活性,降低呼吸链复合物Ⅱ的Vmax,进而减少线粒体ATP的合成量。
胺碘酮是常用抗心律失常药物,Serviddio等[15]发现线粒体氧化应激和对线粒体呼吸链的功能损伤是胺碘酮导致肝毒性的主要原因。胺碘酮诱导肝细胞线粒体功能障碍而发生肝损伤的机制之一是其抑制呼吸链复合物Ⅰ,阻断呼吸链中电子的传递,最终降低ATP的生成。
他莫昔芬在临床上主要用于乳腺癌的治疗,经研究发现线粒体损伤是其诱导肝毒性发生的主要原因。其对肝细胞线粒体的损伤也是多方面的,包括对电子呼吸链的阻断。Ribeiro等[16]发现,他莫昔芬对电子呼吸链的阻断主要通过对呼吸链复合物Ⅰ、Ⅲ、Ⅳ的抑制而发挥作用。Cardoso等[17]研究发现,他莫昔芬及其代谢物4-羟三苯氧胺能通过诱导线粒体膜电位去极化、阻断线粒体的呼吸等作用诱发对大鼠肝脏的毒性。
呋塞米肝毒性非常少见,其所致的肝毒性多数为肝细胞型,研究发现细胞内还原型谷胱甘肽或线粒体内总谷胱甘肽缺乏导致呼吸链出现异常是其产生肝损伤的重要原因之一。
异烟肼的不良反应之一为诱导肝细胞的凋亡,Lee等[18]在对小鼠肝细胞的研究中发现,异烟肼诱导的肝细胞凋亡主要是通过其对线粒体呼吸链复合物Ⅰ的抑制而发挥作用。
中草药雷公藤对细胞免疫和体液免疫均有抑制作用,其主要的有效成分和毒性成分为雷公藤甲素。张陆勇课题组[19-20]研究雷公藤甲素导致SD大鼠肝损伤时发现,雷公藤甲素引起肝损伤与直接损害肝线粒体的结构和功能有关,线粒体损伤可能是雷公藤甲素致肝损伤的机制之一。主要体现在肝细胞线粒体呼吸链的电子传递障碍,线粒体膜电位降低,显著地抑制线粒体呼吸链复合物Ⅰ、Ⅳ等。
除了上述药物,作用于肝细胞线粒体的电子呼吸链而诱发肝损伤的药物,还有中枢神经系统用药阿吡坦和双硫仑、麻醉药物丁丙诺啡、抗肿瘤药物尼鲁米特、循环系统用药哌克昔林、降血糖药物曲格列酮等。
3.2 氧化磷酸化的解偶联
在完整的线粒体内,电子传递与磷酸化是紧密偶联在一起的,当某些化学物质作用于线粒体而导致电子传递与ATP的形成这2个过程分开时,则只进行电子传递而不能生成ATP。解偶联能增大线粒体内膜对H+的通透性,消除H+梯度,抑制电子传递链的偶联以及ADP的磷酸化进程而无ATP生成,从而影响线粒体能量代谢作用[21]。
李妍等[22]在研究线粒体功能及药物分子骨架结构在非甾体抗炎药引起肝损伤中的作用时发现,二苯胺结构的非甾体抗炎药如双氯芬酸钠、托灭酸能导致分离线粒体肿胀,膜电位下降,而环孢素A能明显抑制这一过程。在原代培养肝细胞上,二苯胺结构的非甾体抗炎药能降低细胞ATP含量和下调线粒体膜电位△Ψm。Masubuchi等[23]提取染毒后Wistar大鼠肝脏线粒体进行实验,发现二苯胺作用后的肝线粒体肿胀,MPT孔开放,△Ψm降低,ATP生成锐减,导致能量代谢无法顺利进行从而引发肝损伤。此外,二苯胺还刺激基础氧消耗(Ⅳ态呼吸,ST4)并抑制ADP呼吸(Ⅲ态呼吸,ST3),说明其是线粒体氧化磷酸化的解偶联剂,这也是二苯胺引发能量代谢障碍的主要机制。同时,作为解偶联剂,二苯胺还可激活ATPase,使得ATP水解增多进而耗竭。
Felser等[24]对中枢神经系统用药苯溴马隆诱发肝细胞毒性的研究发现,苯溴马隆主要通过改变肝细胞线粒体的结构和功能产生毒性。在HepG2细胞中,当给予50 µmol·L-1的苯溴马隆24 h后,就出现线粒体解偶联的现象。Simon[25]研究发现,肝细胞线粒体内谷胱甘肽水平的降低进而线粒体呼吸链受到抑制是苯溴马隆诱导肝毒性产生的主要原因。
3.3 直接抑制线粒体脂肪酸β-氧化
对线粒体脂肪酸氧化的抑制主要通过诱发脂肪酸氧化酶活性缺陷、耗竭氧化过程中的L-酰基肉毒碱和辅酶A而发挥作用。
丙戊酸是一种广谱抗惊厥药,其最令人关注的不良反应甚至出现死亡的原因是肝损伤。临床和实验室研究都发现丙戊酸所致的肝损伤涉及免疫系统,然而越来越多的研究发现该药所致的肝损伤主要是由其干扰内源性脂肪酸的β-氧化造成的。有证据提示,丙戊酸硫酯衍生物和辅酶A可能作为代谢中间体存在于肝组织内。肉碱是脂肪酸线粒体β-氧化的重要辅助因子,丙戊酸通过消耗肉碱,减少辅酶A供应和抑制β-氧化的三大功能酶,从而阻碍线粒体β-氧化。此外,丙戊酸经β-氧化生成数种产物,在此过程中,丙戊酸与内源性脂质竞争相关的酶,从而使原有的线粒体潜在的功能缺陷凸显,这些都将诱导肝损伤的发生。Luis等[26]在对丙戊酸诱发肝毒性的研究中也发现,丙戊酸能下调异戊酰辅酶A脱氢酶和2-甲基-3-羟丁酰辅酶A脱氢酶的活性。
3.4 干扰线粒体钙稳态和促进mPTP的开放
线粒体内钙超载可引起氧化磷酸化过程障碍,主要表现在3个方面。其一,胞浆Ca2+水平升高引起线粒体Ca2+由Ca2+单转运蛋白摄取增加而升高。膜电位△Ψm是由于线粒体内膜的质子不均匀分布而产生的,线粒体膜间隙中的质子经复合物Ⅴ-F0顺浓度差返回线粒体基质,同时将储存在线粒体△Ψm中的势能释放,使复合物Ⅴ-F1催化ADP磷酸化生成ATP,即氧化磷酸化。Ca2+单转运蛋白像ATP合酶一样,利用膜电位△Ψm作为驱动力,因此线粒体Ca2+摄取使△Ψm消失,从而诱发氧化磷酸化解偶联的发生,导致ATP合成的减少;此外,作用于NADH的药物使其被氧化时,可激活从线粒体基质间腔逐出Ca2+的转运蛋白,Ca2+由线粒体继续摄取和外运,进一步危害氧化磷酸化过程。其二,线粒体的持续Ca2+累积会产生ROS,线粒体的电子传输链会持续产生超氧化自由基,造成线粒体内膜的氧化损伤,影响ATP合成。其三,胞浆Ca2+的持续升高不仅影响ATP合成,而且也由于Ca2+-ATP酶用于排除多余的Ca2+而增加ATP的消耗。
mPTP是由线粒体内外膜跨膜蛋白共同形成的一种高电导非选择性通道,其生理作用为参与细胞内钙稳态调控、ATP/ADP的转运与能量代谢等,mPTP开放可导致△Ψm下降或消失、氧化磷酸化的解偶联等[27]。线粒体内膜通透性转变是细胞凋亡的必要条件,mPTP打开之后导致线粒体许多功能发生变化并可启动死亡路径。在mPTP开放时线粒体释放细胞凋亡诱导因子(AIF),而从线粒体释放的Cytc也是一种重要的细胞凋亡诱导因子,可与细胞凋亡诱导因子-1(AIF-1)结合并激活,继而使半胱氨酸天冬氨酸蛋白酶-9(caspase-9)激活,被激活的caspase-9能激活其他的半胱氨酸天冬氨酸蛋白酶如caspase-3等,从而诱导细胞凋亡。
对乙酰氨基酚诱导线粒体损伤的机制研究显示,对乙酰氨基酚摄入体内后,其代谢物与肝细胞蛋白巯基共价结合使胞质中钙离子升高,胞浆膜损伤和肝细胞线粒体钙丢失,诱发mPTP通道改变[27]。
非甾体类抗炎药尼美舒利为选择性环氧化酶-2(COX-2)抑制剂,近年来,由于其可引起肝脏损伤,甚至导致死亡。目前只有法国、瑞士、墨西哥等50多个国家在使用该药,而在美国、爱尔兰、新加坡等国家其被禁止销售。Mingatto等[28]通过体外试验认为尼美舒利可通过干扰线粒体的呼吸链,导致细胞内ATP衰竭。此外,尼美舒利可引起线粒体通透性改变,并直接造成线粒体的肿胀和破裂。
抗结核药物异烟肼在体内被代谢为乙酰肼(acethydrazide,AHZ)和肼(hydrazine,HZ),两者均与异烟肼的肝毒性有关,是引起肝损伤的重要毒性代谢产物。异烟肼在肝内经P450酶还原后,产生亲电子基、自由基、氧基等毒性代谢产物。这些代谢产物通过与肝细胞蛋白半胱氨酸残基的巯基、赖氨酸残基的氨基等亲电子基团共价结合,破坏肝细胞膜的完整性和膜的Ca2+-ATP酶系,使细胞内外环境Ca2+的稳态被破坏,肝化学毒性损害时通过Ca2+浓度增加并催化相关酶形成,最后通过共同通路导致肝细胞凋亡[29]。Chowdhury等[30]研究表明,抗结核药物异烟肼和利福平诱导的肝损伤主要与线粒体氧化应激和线粒体mPTP不正确的开放有关。
曲格列酮(troglitazone)作为首个噻唑烷类抗糖尿病药物,上市仅3年就因严重的肝脏毒性被美国食品与药品监督管理局(FDA)禁用而撤市,进一步研究发现其引发肝脏毒性的主要机制是诱导mPTP的开放,导致△Ψm降低和Ca2+蓄积而引起细胞渗透性损伤,引发能量代谢严重障碍[31]。
线粒体是疏水性胆汁酸诱导肝细胞毒性的靶器官之一。Sola等[32]发现在肝细胞水平,胆汁酸主要通过激活caspase引发Cytc释放增加而发挥促凋亡作用;Schulz等[33]在肝细胞水平上发现,甘氨鹅去氧胆酸可与线粒体外膜结合,从而使线粒体内膜失去完整性,导致mMPT打开,最终导致肝细胞凋亡。Barrasa等[34]在人结肠癌细胞上发现,胆汁酸主要通过上调Bcl-2和使Bax失活而诱导细胞凋亡。
3.5 导致线粒体DNA突变
线粒体能够独立进行基因的转录、翻译和蛋白质的表达。线粒体DNA(mitochondrial DNA,mtDNA)为闭合双链DNA分子。人类mtDNA仅编码13种与呼吸功能有关的蛋白质,因此mtDNA的损伤可直接反映线粒体呼吸功能受损。mtDNA无内含子并暴露于呼吸链产生的高水平活性氧的环境下,同时缺少组蛋白的保护,线粒体中又无DNA损伤修复系统,其突变频率较核基因组高10倍以上,且mtDNA序列微小的改变可涉及重要结构基因的变化,导致线粒体功能障碍[35]。mtDNA的损伤先于ATP的耗竭、电子传递的障碍以及线粒体膜电位的崩溃发生,mtDNA一旦受损,氧化磷酸化中ETC关键蛋白的表达受到影响,导致严重的能量代谢障碍[36]。
核苷类及单核苷酸类逆转录酶抑制剂(NRTI和NtRTI)分子中3′ -羟基缺乏可导致HIV合成新DNA链的终止。这些药物治疗时特别在中长期应用时的主要毒性是由于线粒体DNA聚合酶γ受抑,导致线粒体酶合成受损,而线粒体酶是通过氧化磷酸化产生ATP的,同时其在mtDNA复制中也起到关键作用[37]。NRTIs对病毒反转录酶有着高度的免疫亲和力,因此,可有效地抑制HIV的复制。NRTIs也可以和人类DNA聚合酶结合,如DNA聚合酶β和线粒体DNA聚合酶γ,能通过中止DNA复制,使线粒体DNA链复制可逆性中止,减少线粒体DNA含量引起线粒体损伤[38]。其主要机制是造成线粒体DNA中氧化磷酸化系统破坏,导致ATP产生减少,从而引起线粒体DNA损害,进而发生一系列更严重的氧化性损伤和脂质过氧化反应。线粒体毒性由核苷类药物进入细胞的浓度决定,高浓度可导致更显著的mtDNA下降。不同药物的线粒体毒性有显著差别,目前应用于临床的NRTIs中,对线粒体DNA聚合酶γ都有不同的亲和力,会引起不同的毒性。Geddes等[39]的体外实验研究结果证实不同的NRTIs抑制聚合酶γ的强度顺序如下:扎西他滨(ddC)>去羟肌苷(ddI)>司坦夫定(d4T)≥齐多夫定(ZDV)>替诺福韦(TDF)=拉米夫定(3TC)=恩曲他滨(FTC)=阿巴卡韦(ABC)。
综上,肝毒性药物对线粒体的损伤包括结构改变和功能紊乱,其中涉及多种机制(见表1)[40]。任何一种药物对线粒体的损伤都是相互交叉、相互关联的多途径诱导,包括线粒体DNA的复制和表达受到抑制、内膜通透性增加、电子传递体系的抑制和新陈代谢的抑制等,是一个复杂的网络毒性作用。这就要求我们在研究肝毒性药物对线粒体的损伤作用时,应该以一种整体的科研思路进行研究。

表1 肝毒性药物与肝细胞线粒体损伤的相互作用关系Table 1 Hepatotoxic drugs and their corresponding deleterious effects on mitochondrial function and genome
4 肝毒性药物诱发线粒体损伤的生物标志物
4.1 血清乳酸盐
线粒体受损时,细胞由于产生ATP的能力下降而加快糖酵解的速度,从而导致乳酸产生的增加。乳酸中毒后导致的血清乳酸盐升高是药物性线粒体损伤的主要生物标志物[41]。
4.2 核编码蛋白及线粒体DNA编码蛋白比值
渐进型的线粒体损伤是由于基因的表达或线粒体的复制被慢性抑制而产生的,可以通过相对的蛋白量来检测[42]。例如,通过分子生物学技术检测血样,可测定核编码和线粒体DNA编码蛋白的比值,从而检测线粒体能量的缺失。
4.3 线粒体耗氧量、二氧化碳产生量及热能的检测
线粒体功能的损伤也可通过检测线粒体耗氧量以及二氧化碳和热能的产生量来指示。Grattagliano等[43]采用13C-呼吸测试对肝脏线粒体功能进行临床研究,主要包括13C-底物的摄入、十二指肠和空肠吸收、肝脏线粒体代谢、经肺部呼出体外并对13CO2/12CO2进行检测等4个步骤,通过最终13CO2/12CO2比值的变化,间接地反映肝脏线粒体的损伤(见图2)。
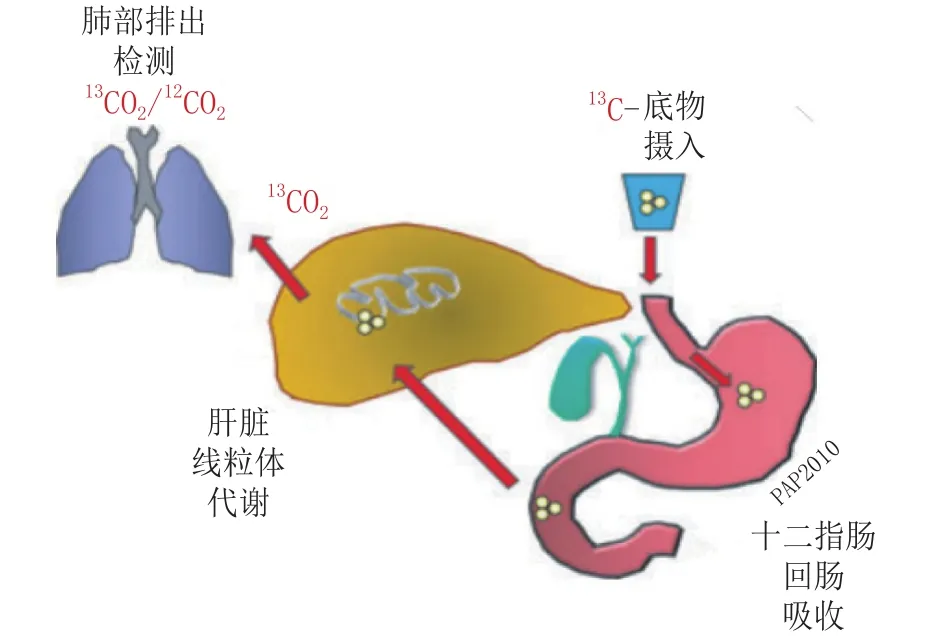
图2 口服13C-呼吸测试检测肝脏线粒体功能的基本原理Figure 2 General principles of oral13C-breath test for liver mitochondrial function
4.4 NADH/NAD比值
NADH是线粒体中能量产生链中的标志物,其水平的上升指示代谢紊乱的出现。NADH/NAD的氧化还原状态是表征细胞内线粒体功能的优选参数[44]。
4.5 自由基的检测
自由基是线粒体在电子传递时产生的。生理条件下,机体自由基的清除系统可使自由基的产生与消除保持在一个相对动态平衡的状态;当线粒体损伤时,体内自由基蓄积过量,过量的自由基损害细胞膜上的脂类和蛋白质,加重自由基的产生,最终导致细胞的死亡。机体内清除自由基的酶系统主要有超氧化物歧化酶SOD、过氧化氢酶CAT、还原型谷胱甘肽酶等酶类组成,可以通过对这些酶活性的检测间接反映线粒体的功能。
4.6 鸟氨酸氨甲酰转移酶的检测
线粒体鸟氨酸氨甲酰转移酶(ornithine carbamyltransferase, OCT)主要存在于肝细胞线粒体中,是肝脏特异性酶,其含量约占血清中OCT的99%。肝细胞受损时,该酶可作为监测肝细胞线粒体损伤的特异性指标[45]。
5 线粒体毒性评价的指标与技术
药物的安全性是决定创新药物研发成败的关键因素之一。随着线粒体功能的逐步阐明,研究人员发现多种药物的作用靶点位于线粒体膜或线粒体内的酶体复合物。线粒体毒性是多种药物研发失败或临床受限的重要原因。线粒体作为许多药物毒性作用的重要靶标,越来越受到国内外药物研发机构、制药公司与管理部门的广泛关注和高度重视。一些国际大型制药公司及药物研发机构纷纷将线粒体毒性评价作为候选药物临床前安全性评价的重要内容。针对药物致线粒体损伤的特点及机制,可从以下几个阶段开展药物线粒体毒性的评价。
5.1 药物早期发现阶段
基于大量候选药物的评价需求,快速、灵敏的高通量筛选、高内涵筛选以及活细胞成像技术成为候选药物早期线粒体毒性评价的重要技术手段[46-47]。
5.1.1 检测指标 这一阶段主要应用体外培养的细胞或分离的线粒体进行试验,开展高通量的线粒体毒性筛选。评价指标包括:细胞的存活率、细胞内ATP含量水平、线粒体耗氧量、线粒体膜电位、膜磷脂、mPTP,细胞内与线粒体内的ROS、线粒体内mtDNA的损伤程度等。
5.1.2 检测技术 mPTP的检测方法有活性物质标记法、膜片钳法、分光光度法等,其中分光光度法较为简单易用。线粒体膜磷脂的检测可先用差速离心法分离线粒体膜磷脂,然后使用高效液相进行检测,也可采用毛细血管电泳联合荧光显微镜技术检测。线粒体膜电位可采用荧光探针的方法进行测定。DNA测序是检测mtDNA序列多态性最常用的方法之一,该方法具有准确度高、结果稳定等特点,也可以使用序列特异性寡核苷酸探针,通过对基因特异性的寡核苷酸探针与基因的杂交情况,判定mtDNA是否发生了基因突变[48]。
5.2 药物发现阶段或临床前实验过程
为了更加灵敏地发现候选药物的线粒体毒性风险,近年来选择合适的动物模型进行线粒体毒性评价成为一个重要发展方向。如有研究表明,锰超氧化物歧化酶部分敲除(MnSOD+/-)小鼠、线粒体转录因子A部分敲除(TFAM+/-)小鼠以及金属硫蛋白敲除(MT-/-)小鼠对药物诱导的靶器官线粒体毒性更为敏感[46,49-50],提示这些转基因动物模型在候选药物线粒体毒性筛选以及线粒体损伤相关机制研究中具有重要的应用价值。
抗病毒核苷类药物是目前临床上治疗肝炎、艾滋病等病毒性疾病的首选药物,然而此类药物可通过干扰mtDNA合成产生严重的线粒体毒性。缺乏合适的线粒体毒性评价模型是限制我国开展抗病毒核苷类药物研发的主要技术瓶颈。张陆勇课题组[51-53]在全国率先建立了以我国特有种属喜马拉雅旱獭为动物模型的药物线粒体毒性评价新模型,并结合HepG2细胞体外线粒体毒性评价模型,形成较完善的核苷类药物体内外线粒体毒性评价体系。同时,曾文等研究者[54-56]在国内建立了以恒河猴为动物模型的药物体内线粒体毒性评价方法,并应用该模型评价了核苷类药物美他卡韦(metacavir)的线粒体毒性。这些新方法为核苷类抗病毒药物线粒体毒性的科学评价提供了技术支撑,推进了该类药物的研发进程。
5.3 药物在人体中致靶器官线粒体毒性的预测研究阶段
随着药物筛选、计算生物学技术、计算机毒理学和现代化组学技术的发展,近年来还发展起来一种基于特定靶点和定量构效关系分析的虚拟筛选技术,并应用于线粒体毒性筛选[57]。
6 结语
保护线粒体结构和功能的完整性不但对肝细胞的生存起着重要作用,还能保证为细胞提供足够的能量。虽然药源性肝损伤的作用机制非常广泛,但线粒体是其最容易被攻击的靶器官,线粒体损伤在药源性肝损伤中的作用越来越受到重视。众多研究也表明,线粒体功能障碍是药物诱发肝损伤的主要机制。药源性肝损伤防治的关键在于预防和合理用药,尽量避免使用或大剂量使用容易致肝损伤的药物和及早进行监测预警。因此,我们仍需做更多的调查和临床观察,以确定能诱导肝细胞线粒体损伤并导致肝脏病变的药物列表,对药源性肝损伤做到早发现、早预防和早治疗。然而,许多肝毒性药物对线粒体的损伤是多途径、多方位的,故应从整体的、系统的视角来进行线粒体功能障碍所致药源性肝损伤的研究。随着人们对线粒体结构和功能的基础理论研究的深入,结合基因组学、蛋白组学、代谢组学技术和虚拟筛选技术等现代技术的应用,越来越多新的肝细胞线粒体毒性的潜在生物标志物被发现,并预计将来有可能应用于肝毒性的早期预测。同时,通过提高对药源性肝损伤的诊断水平,加强因果关系评估和临床风险分析,也能使药物毒理学更好地指导临床安全、合理用药。
[1]Guengerich F P. Mechanisms of drug toxicity and relevance to pharmaceutical development[J].Drug Meta Pharmacokinet,2011,26(1):3-14.
[2]陈成伟.药物与中毒性肝病(2版)[M].上海:上海科学技术出版社,2012:3-5.
[3]张蕾,于峰.肝细胞线粒体凋亡途径及保护机制研究进展[J].现代生物医学进展,2014,14(3):586-589.
[4]熊燕,张梅,陈菲,等.线粒体功能障碍与心血管疾病[J].中国病理生理杂志,2013,29(2):364-370.
[5]EI-Khoury T G , Bahr G M , Echtay K S. Muramyl-dipeptideinduced mitochondrial proton leak in macrophages is associated with upregulation of uncoupling protein 2 and the production of reactive oxygen and reactive nitrogen species[J]. FEBS J,2011,278(17):3054-3064.
[6]Tarasov A I, Griffiths E J, Rutter G A. Regulation of ATP production by mitochondrial Ca2+[J]. Cell Calcium,2012,52(1):28-35.
[7]Sheng B, Wang X, Su B, et al. Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer′s disease [J]. JNeurochem,2012,120(3):419-429.
[8]Shokolenko I, Venediktova N, Bochkareva A, et al. Oxidative stress induces degradation of mitochondrial DNA[J]. Nucleic Acids Res,2009,37(8):2539-2548.
[9]Dykens J A, Will Y. The significance of mitochondrial toxicity testing in drug development[J]. Drug Discov Today,2007,12(17/18):777-785.
[10]肖芳,钟才高.外源化学为干扰肝细胞线粒体ATP合成体系机制的研究进展[J].中国药理学与毒理学杂志,2010,24(3):232-235.
[11]Tai Y K, Cheong Y M,Almsherqi Z A,et al. High dose Clopidogrel decreases mice liver mitochondrial respiration function in vitro[J]. Int J Cardiol,2009,133(2):250-252.
[12]Zahno A, Bouitbir J, Maseneni S, et al. Hepatocellular toxicity of clopidogrel: mechanisms and risk factors[J]. Free Radic Biol Med,2013,65:208-216.
[13]潘家琪,宋丹军,李鹏旭,等.对乙酰氨基酚肝毒性机制与治疗研究进展[J].中国药理学与毒理学杂志,2014,28(4):618-624.
[14]Kaushal R, Dave K R, Katyare S S, et al. Paracetamol hepatotoxicity and microsomal function[J]. Environ Toxicol Pharmacol,1999,7(1):67-74.
[15]Serviddio G, Bellanti F, Giudetti A M, et al. Mitochondrial oxidative stress and respiratory chain dysfunction account for liver toxicity during amiodarone but not dronedarone administration[J]. Free Radic Biol Med,2011,51(12):2234-2242.
[16]Ribeiro M P, Santos A E, Custódio J B, et al. Mitochondria: the gateway for tamoxifen-induced liver injury[J]. Toxicology,2014,323:10-18.
[17]Cardoso C M, Moreno A J, Almeida L M, et al. Comparison of the changes in adenine nucleotides of rat liver mitochondria induced by tamoxifen and 4-hydroxytamoxifen[J].Toxicology in Vitro,2003,17(5/6):663-670.
[18]Lee K K, Fujimoto K, Zhang C, et al. Isoniazid-induced cell death is precipitated by underlying mitochondrial complex I dysfunction in mouse hepatocytes[J]. Free Radic Biol Med,2013,65:584-594.
[19]Fu Q, Huang X, Shu B, et al. Inhibition of mitochondrial respiratory chain is involved in triptolide-induced liver injury[J]. Fitoterapia,2011,82(8):1241-1248.
[20]FU Q, Jiang Z, Zhang L. Impairment of triptolide on liver mitochondrial in isolated liver mitochondria and HL7702 cell line[J]. Chin J Integr Med,2012,18(11):1-5.
[21]Toyamizu M, Okamoto K, Ishibashi T, et al.Uncoupling effect of anacardic acids from cashew nutshell oil on oxidative phosphorylation of rat liver mitochondrial [J].Life Sci,2000,66(3):229-234.
[22]李妍,任进.线粒体损伤和二苯胺结构在非甾体类抗炎药引起的肝细胞损伤中的作用[C]//2008中国药学会学术年会暨第八届中国药师周论文.石家庄:中国药学会,2008.
[23]Masubuchi Y, Yamada S,Horie T,et al. Possible mechanism of hepatocyte injury induced by diphenylamine and its structurally related nonsteroidal anti-inflammatory drugs[J]. J Pharmcol Exp Ther,2000,292(3):982-987.
[24]Felser A, Lindinger P W, Schnell D, et al. Hepatocellular toxicity of benzbromarone: effects on mitochondrial function and structure[J]. Toxicology,2014,324:136-146.
[25]Qu Q, Liu J, Zhou H H,et al. Nrf2 protects against furosemide-induced hepatotoxicity[J].Toxicology,2014,324:35-42.
[26]Luis P. Interaction of VPA metabolites with branched-chain amino acids oxidation: implications for drug-hepatotoxicity[J].Toxicol Lett,2008,180S: S32-S246.
[27]Trost L C, Lemasters J J. Role of the mitochondrial permeability transition in salicylate toxicity to cultured rat hepatocytes: implications for the pathogenesis of Reye’s syndrome[J]. Toxicol Appl Pharmacol,1997,147(2):431-441.
[28]Mingatto F B, Rodrigues T, Pigoso A A,et al.The critical role of mitochondrial energetic impaiment in the toxicity of nimesulide to hepatocytes[J]. J Pharmaco Exp Ther,2002,303(2):601-607.
[29]Desmet V J. Milestones in liver disease: scoring chronic hepatitis[J]. J Hepatol,2003,38(4):382-386.
[30]Chowdhury A, Santra A, Bhattacharjee K, et al. Mitochondrial oxidative stress and permeability transition in Isoniazid and Rifampicin induced liver injury in mice[J]. J Hepatol,2006,45(1):117-126.
[31]Masubuchi Y, Kano S, Horie T, et al. Mitochondrial permeabitily transition as a potential determinant of hepatotoxicity of antidiabetic thiazolidinediones[J].Toxicology,2006,222(3):233-239.
[32]Sola S, Amaral J D, Aranha M M, et al. modulation of hepatocyte apoptosis:cross-talk between bile acids and nuclear steroid receptors [J]. Curt Med Chem,2006,13(25):3039-3051.
[33]Schulz S, Schmitt S, Wimmer R,et al. Progressive stages of mitochondrial destruction caused by cell toxic bile salts [J]. Biochimica et Biophysica Acta,2013,1828(9):2121-2133.
[34]Barrasa J I, Santiago-Gómez A, Olmo N, et al. Resistance to butyrate impairs bile acid-induced apoptosis in human colon adenocarcinoma
[46]Nadanaciva S, Diliman K, Gebhard D F, et al . High-content screening for compounds that affect mtDNA-encoded protein levels in eukaryotic cells[J]. J Biomol Screen,2010,15(8):937-948.
[47]Schoonen W G, Westerink W M, Horbach G J. High-throughput screening for analysis of in vitro toxicity[J]. EXS,2009,99:401-452.
[48]左钱飞,张海献,鲁鹏飞.线粒体损伤与检测方法研究进展[J].科学之友,2009,06(17):5-6.
[49]Koczor C, Kohler J, Lewis W. Transgenic mouse models of mitochondrial toxicity associated with HIV/AIDS and antiretrovirals[J]. Methods,2010,51(4):399-404.
[50]Fu Z, Guo J, Jing L,et al. Enhanced toxicity and ROS generation by doxorubicin in primary cultures of cardiomyocytes from neonatal metallothionein-I/II null mice[J]. Toxicol in Vitro,2010,24(6):1584-1591.
[51]Zhang P, Zhang L, Jiang Z, et al. In vitro mitochondrial toxicity of metacavir, a new nucleoside reverse transcriptase inhibitor for treatment of hepatitis B virus[J]. Antimicrob Agents Chemother,2010,54(11):4887-4892.
[52]Zhang P, Zhang L, Jiang Z, et al. evaluation of mitochondrial toxicity in marmota himalayana treated with metacavir, a novel 2′,3′- dideoxyguanosine prodrug for treatment of hepatitis B virus[J]. Antimicrob Agents Chemother,2011,55(5):1930-1936.
[53]张评浒,陶元清,江振洲,等.喜马拉雅旱獭作为药物线粒体毒性替代模型的可行性分析[J].实验动物与比较医学,2012,32(5):436-440.
[54]Zeng W, Cheng AC, Chen ZL, et al. In vivo assessment of mitochondrial toxicity of metacavir in Rhesus monkeys after three months of intravenous administration[J]. Acta Pharmacologica Sinica,2009,30(12):1666-1673.
[55]杨遵远,龚立,冯兴磊,等.采用HepG2和HK-2细胞模型评价富马酸泰诺福韦双特戊酯的线粒体毒性[J].中国新药杂志,2013,22(5):513-519.
[56]龚立,曾文,申渝波,等.采用HepG2细胞模型评价BP0018的线粒体毒性[C]//首届中国药物毒理学年会(2011年)暨国际药物非临床安全性评价研究论坛论文集.上海:中国毒理学会药物毒理与安全性评价专业委员会,2011.
[57]Marella M, Seo B B, Thpmas B B, et al. Successful amelioration of mitochondrial optic neuropathy using the yeast NDI1 gene in a rat animal model[J].2010,5(7): e11472. cells via up-regulation of Bcl-2 and inactivation of Bax [J]. Biochimica et Biophysica Acta,2012,1823(12):2201-2209.
[35]Higuchi M. Regulation of mitochondrial DNA content and cancer [J]. Mitochondrial,2007,7(1-2):53-57.
[36]Van Houten B, Woshner V, Santos J H. Role of mitochondrial DNA in toxic responses to oxidative stress[J]. DNA Repair,2006,5(2):145-152.
[37]郑方算,黄涛.抗病毒治疗的不良反应[J].国外医药(合成药生化药制剂分册),2001,22(5):310-313.
[38]Walker U A,Bickel M, Luke V S, et al . Evidence of nucleoside analogue reverse transcriptase inhibitor associated genetic and structural defects of mitochondrial in adipose tissue of HIV-infected patients[J].J Acquire Immune Deficsyndr,2002,29(2):117-121.
[39]Geddes R, Knight S, Moosa M Y, et al. A high incidence of nucleoside reverse transcriptase inhibitor(NRTI)-induced lactic acidosis in HIV-infected patients in a South African context[J]. S Afr Med J,2006,96(8):722-724.
[40]Begriche K, Massart J, Robin MA,et al. Drug-induced toxicity on mitochondria and lipid metabolism: mechanistic diversity and deleterious consequences for the liver[J]. J Hepatol,2011,54(4):773-794.
[41]Mancuso M, Orsucci D,Coppede F, et al. Diagnostic approach to mitochondrial disorders: the need for a reliable biomarker[J]. Curr Mol Med,2009,9(9):1095-1107.
[42]Shikuma C M, Gerschenson M, Chow D, et al. Mitochonrial oxidative phosphorylation protein levels in peripheral blood mononuclear cells correlate with levels in subcutaneous adipose tissue within samples differing by HIV and lipoatrrophy status[J]. Aids Res Hum Retroviruses,2008,24(10):1255-1262.
[43]Grattagliano I, Lauterburg B H, Palasciano G, et al.13C-breath tests for clinical investigation of liver mitochondrial function[J]. Eur J Clin Invest,2010,40(9):843-850.
[44]Loizzo S, Pieri M, Ferri A,et al. Dynamic DAN(P)H post-synaptic
autofluorescence signals for the assessment of mitochondiral function
in a neurodegenerative disease: monitoring the primary motor cortex of G93A mice, an amyotrophic lateral sclerosis model[J]. Mitochondrial,2010,10(2):108-114.
[45]刘晓娜,刘密凤,贾栗,等.药源性肝损伤潜在血清生物标志物的研究进展[J].首都医科大学学报,2014,35(4):511-515.
Research Progress in Drug-induced Liver Injury via Mitochondrial Damage
YANG Tingting1,2, JIANG Zhenzhou1,3, ZHANG Luyong1,2
( 1.Jiangsu Key Laboratory of Drug Screening, China Pharmaceutical University, Nanjing 210009, China; 2. Jiangsu Center for Pharmacodynamics Research and Evaluation, China Pharmaceutical University, Nanjing 210009, China; 3. Key Laboratory of Drug Quality Control and Pharmacovigilance, China Pharmaceutical University, Ministry of Education, Nanjing 210009, China)
Mitochondria are major organelles of energy generation and material oxidation in cell and play a fundamental role in energy metabolism, free radicals production, aging, and apoptotic regulation.The possible factors leading to mitochondrial dysfunction include respiratory chain defects, metabolic enzyme inactivation, structural changes, mutations, etc. All of which will affect the normal function of the cells, resulting in the occurance of diseases. Mitochondria are important targets of drug toxicity, and the liver is also a major target of drug damage because it is an important organ of drug metabolism. A variety of clinical drugs, such as antivirals, anti-cancer drugs and antibiotics have significantly shown the inducible mitochondrial injury of liver.Drugs induced liver injury primarily by changing the activity of enzyme and the structure of mitochondrial and/or decreasing the synthesis of mtDNA, further undermining β-lipid oxidation and the oxidative of liver cells.This paper provides the overview of research progress on the role of mitochondrial damage in drug-induced liver injury and gives thoughts on the prediction and prevention of drug-induced liver injury.
mitochondrial; mitochondrial damage; drug-induced liver injury; hepetotoxicity; clinical drug
R969
A
1001-5094(2014)11-0809-10
接受日期:2014-10-20
项目资助: 国家自然科学基金重大国际(地区)合作研究项目(No. 81320108029);国家自然科学基金面上项目(No.81273604);国家自然科学基金面上项目(No.81173651)
*通讯作者:江振洲,副研究员;
研究方向:分子药理学和毒代动力学;
Tel: 025-83271043; E-mail: beaglejiang@cpu.edu.cn
