抗多药耐药靶点及相关药物的研究进展
2014-03-09吴晨徐竹轩周启蒙支运宝林克江
吴晨,徐竹轩,周启蒙,支运宝,林克江
(中国药科大学药学院,江苏 南京 210009)
抗多药耐药靶点及相关药物的研究进展
吴晨,徐竹轩,周启蒙,支运宝,林克江*
(中国药科大学药学院,江苏 南京 210009)
抗多药耐药是指在疾病的治疗过程中,细胞对多种药物产生广泛的耐受,导致治疗效果不理想的现象。多药耐药多发于感染与肿瘤疾病的治疗中,已成为治愈这2类疾病的主要障碍。从转运蛋白和离子通道、酶以及核糖体这3个方面综述抗多药耐药靶点的机制及相关药物的研究进展,旨在为抗多药耐药药物的研发提供参考。
多药耐药;感染;肿瘤;转运蛋白;酶;核糖体蛋白
多药耐药(multidrug resistance,MDR)是指在疾病的治疗过程中,细胞对多种药物产生广泛的耐受,导致治疗效果不理想的现象。多药耐药多发于感染与肿瘤疾病治疗中,已成为治愈这2类疾病的重要障碍。目前多药耐药研究的难点在于耐药性基因突变多样性及机制的复杂性,导致药物研发处于停滞状态。因此,有必要对目前已知的多药耐药机制进行总结,为抗多药耐药药物的进一步研发寻找突破口。
目前多药耐药主要机制可归纳为以下2类[1]。1)减少水溶性药物经转运体摄入细胞的量以及增加细胞向外泵出药物的能力。与此相关的主要有转运蛋白和离子通道,其中转运蛋白主要为ATP结合盒(ATP-binding cassette,ABC)转运蛋白超家族,其包括P-糖蛋白(P-gp)、多药耐药相关蛋白1(MRP1)和乳腺癌抗性蛋白(BCRP);离子通道以钾离子通道为主。2)细胞自身发生改变,降低细胞毒性药物的效果,与此相关的功能蛋白,包括影响细胞周期、增加DNA和RNA损伤后修复、减少凋亡和影响药物代谢的酶类[如DNA解旋酶(DNA gyrase)、DNA拓扑异构酶(DNA Topoisomerase)、二氢叶酸合酶(dihydropteroate synthase,dhpS)、烯酰ACP还原酶、β-内酰胺酶(β-lactamase,BL)、蛋白激酶类],以及核糖体及其相关蛋白。此外,还有一些潜在的耐药性靶点如脂蛋白(LPS)、膜黏附蛋白、Toll样受体(TLR)等(见图1)。本文对多药耐药相关靶点及药物的研究进展进行综述,旨在为抗多药耐药药物的进一步研发提供参考。

图1 细菌感染中的多药耐药靶点Figure 1 The multidrug resistant targets in bacterial infections
1 转运蛋白与离子通道
转运蛋白与离子通道,其共同的功能是调控细胞内外物质的运输。不同转运蛋白对运输底物的选择性存在差异,有专一性的,也有非特异性的,转运蛋白功能的异常可降低药物的有效浓度,从而导致耐药,是研究多药耐药的主要靶点。
1.1 ABC转运蛋白超家族
ABC转运蛋白超家族(ATP结合盒转运蛋白,ATP-binding cassette transporters,简称ABC transporters),在多药耐药中占有重要地位。其结构上的功能性区域包括跨膜区(transmembrane domain,TMD)和核苷(如ATP)结合区(nucleotide-binding domain,NBD)[2]。NBD域与ATP结合后,TMD域的螺旋结构打开一个小孔,推动底物向外排出[3]。
人类共有48个基因编码ABC转运蛋白,分为A~G共7个亚族[4]。ABC转运蛋白作用范围广泛,从正常代谢产物到化疗药物均可为其所转运,同时众多ABC互相协作构成复杂的转运网络。ABC转运蛋白有选择性地在刷状肠道细胞、胆汁微管的肝细胞、肾近端小管、血脑屏障的上皮细胞等重要的生理屏障中高度表达,参与多种生理过程,例如脂类代谢、体内离子平衡和免疫功能等(见图2)。引起MDR的ABC转运蛋白集中于ABCB亚族、ABCC亚族、ABCG亚族,具有代表性的成员包括P-糖蛋白(P-glycoprotein,P-gp,ABCB1),多药耐药相关蛋白(multidrug resistance associated-protein 1,MRP1,ABCC1) 以 及 乳 腺 癌抗 性 蛋 白(breast cancer resistance protein,BCRP,ABCG2)。

图2 ABC转运蛋白超家族成员的分布Figure 2 The location of ABC transporters family members
1.1.1 ABCB亚族 ABCB亚族的代表是ABCB1,也常称为P-糖蛋白、P-gp或MDR1。化疗中的主导药物,包括长春花生物碱类、蒽环类药物、表鬼臼毒素和紫杉烷,都能被ABCB1转运。维拉帕米可以抑制ABCB1[5],从而增强此类细胞毒药物对于ABCB1高表达细胞的杀灭效果。
此外该亚族中,ABCB11[6-7]和ABCB4(MDR3)[8-9]在肝脏中表达,前者转运胆盐,后者转运磷脂酰胆碱固醇。人工表达ABCB11,可让细胞获得对紫杉醇的耐药性[10];而ABCB4可以促进地高辛、紫杉醇和长春碱的转运[11]。
目前针对ABCB类研发的药物不多,当前在研的9个药物中8个处于生物活性测试阶段,均为ABCB1抑制剂。其中,有3个药物抑制ABCB1的同时还可抑制ABCG2。唯一1个进入临床前阶段的药物由中国医学科学院研发,具有多重作用机制,包括ABCB1表达抑制作用、自噬诱导作用、组蛋白脱乙酰酶SIRT1抑制作用、细胞凋亡诱导作用、血管生成抑制作用。综上所述,ABCB1是较有开发前景的靶标,ABCB11和ABCB4也有待进一步研究。
1.1.2 ABCC亚族 ABCC亚族蛋白在结构上较ABCB多出一个特征性的链接区(linker region)。ABCC成员可以转运有机阴离子和药物的Ⅱ相代谢产物,与药物Ⅰ相/Ⅱ相代谢的酶系联合作用,为细胞提供了强大的药物消除能力。
ABCC1即MRP1,在多种组织如肿瘤中表达。MRP1以疏水药物为底物,还以谷胱甘肽、葡萄糖醛酸和硫酸结合的药物二次代谢后产物为底物。与ABCC1同源的ABCC2(MRP2)则参与药物的胆汁排泄,常引起口服药物的耐药[12];ABCC3(MRP3)常与葡萄糖醛酸复合物结合[13],可能参与药物的肝肠循环[14];人工转染ABCC6(MRP6)可使细胞产生对依托泊苷、替尼泊苷、阿霉素和柔毛霉素等天然产物的耐药性[15];ABCC10(MRP7)可促使细胞对紫杉醇耐药[16]。
针对肿瘤耐药,以ABCC1为靶点的药物研究较多。现报道的30个药物中,已有环香草酮(cyclovalone)上市,多非喹达(dofequidar)进入Ⅲ期临床研究阶段。也有研究者使用依氟鸟氨酸、舒林酸、奥美拉唑和酮洛芬等早已上市的药物,通过调控ABCC基因,直接从源头调节ABCC蛋白的表达,从而达到抑制耐药性的目的。目前,ABCC1作为抗耐药性靶标,已成为抗肿瘤药研究热点之一。
1.1.3 ABCG亚族 ABCG2(MXR/BCRP)与癌细胞耐药呈明显相关性。ABCG2转运底物包括细胞毒药物,食物中的致癌物及内源性物质[17-18]。ABCG2不仅可以运输甲氨蝶呤,还可转运谷氨酸。对于甲氨蝶呤,ABCG2可产生抗性[19]。ABCG2亦可转运酪氨酸激酶抑制剂等较新品种抗癌药[20]。
目前针对肿瘤耐药,以ABCG2为靶点的药物多达82个,自2011年起至今有4个药物上市,其作用范围较广,涉及不止一个靶标。其余有70余个药物处于生物活性测试阶段,且具有较好的应用前景。
1.1.4 非特异亚型ABC 除了以上介绍的3个亚族外,其他的亚族合称为非特异亚型ABC,其代表为ABCA2。细胞筛选实验表明,ABCA2是导致雌莫司汀和米托蒽醌耐药的主要原因,ABCA2的表达水平与机体对药物的敏感性密切相关。目前对其他亚型的研究还不多,但可以确定的是非特异亚型ABC的表达能够降低肿瘤细胞对药物的敏感性[21]。此外,非特异亚型ABC也与抗菌药物耐药性的产生相关。
本类药物的开发尚不多见:针对原核生物感染,当前有3个药物正在研发中;针对MDR肿瘤,有7个药物处于生物活性测试阶段,有1个进入临床前研究阶段,其中2个药物除了作用于ABC外,还可分别抑制DNA拓扑异构酶Ⅱ和cGMP磷酸二酯酶活性。
1.2 钾离子通道
钾离子通道是生物膜上种类最多,分布最广,最复杂的一类离子通道,多具有ATP依赖性。根据钾离子通道亚单位蛋白质的一级结构,将其划分为3大类:1)含6个跨膜区单孔的电压门控式钾离子通道;2)含2个跨膜区单孔内向整流钾离子通道;3)含4个跨膜区双孔的钾离子通道。钾离子通道通过控制钾泵的开放与关闭改变膜内外离子平衡,进而改变膜电位,引起钙离子相关信号转导通路的改变和相关应激反应。
有研究表明,离子通道蛋白过度表达可能是导致肿瘤治疗效果不佳的原因。已有报道称强心类固醇如cardenolides和bufadienolides以Na+-K+-ATP酶的α亚基为靶点,调节细胞内外K+浓度,改变膜电位从而影响肿瘤细胞的生命活动。强心类固醇药物对非小细胞肺癌、黑色素瘤和肾癌的治疗效果变差与Na+-K+-ATP酶α亚基的过度表达相关,表明钾离子通道相关蛋白是肿瘤细胞耐药性产生的原因之一[22]。
钾离子通道与细菌多药耐药的相关性研究主要在结核分枝杆菌中展开[23]。研究已经确认,钾离子通道抑制剂如维拉帕米、利舍平和乌本(箭毒)苷(ouabain)可以通过抑制吞噬体-溶酶体复合物中离子泵的K+外流,增强杀菌药物的效果,从而改善结核分枝杆菌的多药耐药问题。
目前还没有针对钾离子通道的抗肿瘤多药耐药性的上市药物,相关机制还有待研究。在抗多药耐药菌药物的研究方面,针对钾离子通道的药物达托霉素已于2011年上市,适应证为耐甲氧西林金黄色葡萄球菌(MRSA)引发的感染。离子泵(尤其是钾离子通道)是颇具前景的多药耐药靶点。
2 酶
细菌和肿瘤细胞中酶类的改变也是导致耐药性的重要因素,编码酶的基因发生突变是耐药性产生的根本原因。目前已明确的与耐药性相关的酶包括:DNA解旋酶、DNA拓扑异构酶Ⅳ、二氢叶酸合成酶、烯酰ACP还原酶、β-内酰胺酶和蛋白激酶。
2.1 DNA解旋酶
DNA解旋酶(DNA gyrase)广泛存在于细菌细胞中,其作用是在DNA分子中直接引入负超螺旋。负超螺旋化对DNA复制起始及促进DNA与起始蛋白结合有重要作用。DNA解旋酶由2个亚基——gyrA和gyrB组成,其中gyrA亚基具有DNA结合功能,被认为是氟喹诺酮类药物的靶点,氟喹诺酮类药物通过与解旋酶、DNA生成复合物起效;而gyrB则起到与ATP结合、水解作用,并可被香豆素类化合物抑制[24]。
喹诺酮类药物耐药性产生与DNA解旋酶亚基gyrA和gyrB突变有关。其中gyrA突变概率较gyrB大,主要发生在67至106位氨基酸之间。其中Ser83与Asp87是最常突变的2个位点,为喹诺酮耐药性决定区域(quinolone resistance-determining region,QRDR)。Ser83Trp突变能抑制喹诺酮类药物诺氟沙星与解旋酶-DNA复合物结合。因此,gyrA中QRDR氨基酸改变导致喹诺酮结合位点结构改变,药物与修饰过的酶-DNA复合物亲和力降低,从而产生耐药性。
gyrB突变主要发生在426或447位氨基酸上,即:Asp426Asn 和Lys447Glu。在第一类突变中,426位负电荷消失,这一改变导致喹诺酮药物的疏水正电性基团与位点亲和力下降;第二类突变中447位电荷由正变负,疏水性降低,gyrB与喹诺酮结合能力减弱[25]。
除喹诺酮结合位点的直接突变外,gyrB其他位点的突变与拓扑异构酶Ⅳ ParE突变共同导致耐药性的产生。单位点的突变导致的耐药性较弱,多位点突变是导致耐药性的主要原因,这也是研发抗耐药性药物困难的因素之一[26]。
目前已上市的莫西沙星以DNA解旋酶为靶点,对多种多药耐药的革兰阴性菌有效;GSK-2140944处于临床前研究阶段,主要针对MRSA。可见,DNA解旋酶是对抗多药耐药菌的有效靶点。针对这一靶点研发新广谱抗生素将有助于解决多药耐药问题。
2.2 DNA 拓扑异构酶Ⅳ
细菌DNA合成、mRNA的转录以及细胞分裂都涉及拓扑异构酶催化染色体超螺旋调节机制,包括DNA双链的断裂和重组过程。这些过程被研究人员认为是抗菌药物的靶点。以喹诺酮类药物为例,其以DNA-拓扑异构酶复合物为作用靶点,生成喹诺酮-拓扑异构酶-DNA复合物,通过抑制DNA拓扑异构酶Ⅳ使DNA双链维持在断裂状态并阻碍其重组,从而达到破坏染色体拓扑结构的目的。
拓扑异构酶引发的耐药性机制如图3所示。拓扑异构酶被抑制后细胞产生活性氧(ROS),导致DNA双链断裂并引起DNA应激反应(SOS应答)。在此过程中DNA损伤激活RecA并引起阻遏蛋白lexA自我分裂,从而诱导SOS应答基因表达DNA修复酶,使得拓扑异构酶功能恢复。值得注意的是,几项研究已经表明抑制SOS应答不仅可以提高喹诺酮药效,还可以通过阻碍易出错DNA聚合酶的产生以及耐药性因子的同源重组和横向转移进而减少耐药性突变的发生[27]。
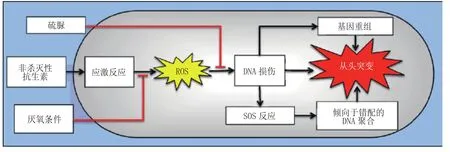
图3 拓扑异构酶引发的多药耐药机制Figure 3 The multidrug resistance mechanism induced by topoisomerase
莫西沙星同时以DNA解旋酶和DNA拓扑异构酶Ⅳ为靶点,对多种多药耐药的革兰阴性菌有效。上述2种酶具有研究前景,新药开发可以同时以这2种酶作为研究对象。
2.3 二氢叶酸合酶
dhpS是细菌叶酸合成的关键酶。磺胺类药物以细菌dhpS为作用靶点。磺胺类药物与dhpS底物——对氨基苯甲酸(PABA)结构相似,可与其竞争性结合dhpS,从而达到抑制dhpS的作用。
磺胺类药物引发耐药性的主要原因为编码dhpS的基因FolP发生突变,导致dhpS结构变化。不同类别的菌株发生突变的位点不同。例如,在大肠杆菌菌株中,dhpS的28位苯丙氨酸(Phe)突变为亮氨酸(Leu);弯曲杆菌有4个氨基酸残基发生突变,突变方式分别为L186F、D238N、N245K和F246Y;而在金黄色葡萄球菌中则有14个氨基酸与耐药性产生相关[28]。
磺胺类药物另一耐药性机制发生在革兰阴性肠道细菌中,由质粒传递基因SUL1和SUL2产生。在SUL1和SUL2基因作用下产生的dhpS能很好地区别PABA与磺胺药。
以dhpS为靶点的药物有复方增效磺胺,目前已进入Ⅲ期临床研究,研究表明其对MRSA引发的感染有效。
2.4 烯酰ACP还原酶
烯酰ACP还原酶有4种同工酶,即FabI、FabK、FabL、FabV,分布于不同种属的细菌中。FabI是细菌合成脂肪酸的关键酶。在催化细菌脂肪酸循环(FASII)的最后一步反应中,反式-2-脂烯酰基ACP的碳碳双键被NADH依赖型FabI酶催化还原,产生酰基-ACP,此酶为脂肪酸合成反应的限速酶。三氯生(TCL)靶向作用于烯酰基底物结合口袋,极大地提高FabI与NAD+的亲和力,并与两者形成稳定的FabI-NAD+-TCL三元复合物。而TCL与酰基底物结合口袋的稳定结合需要FabI与辅因子NAD+相互作用。
TCL耐药性的产生主要由FabI错义突变引起。当93位氨基酸突变(G93V)后,TCL无法继续不可逆结合FabI[G93V]底物结合口袋,同时NAD+与FabI[G93V]结合能力也大大降低。TCL既不可继续作为FabI[G93V]终止抑制剂,也不可与FabI[G93V]、NAD+形成稳定的三元复合物。细菌FabI[G93V]与底物烯酰基ACP恢复结合,脂肪酸恢复合成,细菌从而产生耐药性[29]。
以烯酰ACP还原酶(FabI)为靶点的药物有CG-400549,目前已进入Ⅱ期临床研究,其对MRSA引发的感染有效。
2.5 β-内酰胺酶
β-内酰胺酶能够水解药物分子中的β-内酰胺四元环,作用于临床上广泛使用的一大类以青霉素、头孢菌素为代表的β-内酰胺抗生素,使其水解从而丧失抑制细菌细胞壁合成的作用。随着β-内酰胺抗生素在临床上的广泛使用,编码β-内酰胺酶的基因可发生替换和重组,这些变化可借助质粒等基因载体在不同菌株中传播,引起广泛和强力的耐药现象。β-内酰胺酶突变已成为现今细菌多药耐药的重要原因。
β-内酰胺酶根据一级结构分为A~D四大类。A、C、D类酶催化中心为丝氨酸,B类催化中心则由锌离子构成。与通常位于染色体中的B、C类β-内酰胺酶基因不同,编码A、D类β-内酰胺酶的基因多存在于细菌转座子或质粒上,因此更易于在不同菌株间传播,引起大范围的耐药。A和D类β-内酰胺酶作用底物广泛,对多种抗生素甚至是新型抗生素均有效,也被称为广谱β-内酰胺酶(extended-spectrum β-lactamases,ESBLs)[30]。ESBLs尚无标准化的命名法,常见的类别有:SHV、TEM、OXA、IMP、VIM、KPC等。也有一些罕见的ESBLs,目前只在个别区域的少数菌株中发现。以下仅介绍现今临床影响较为广泛的几种ESBLs。
2.5.1 TEM型ESBLs(A类) 目前确证的TEM多达130种,以TEM-1为代表,在保留酶活性的前提下,其活性中心附近的氨基酸可以发生多种变化[31],这些突变扩大了TEM底物的范围,可引起包括氧基亚氨基β-内酰胺抗生素(oxyimino-β-lactams)在内的抗生素的耐药[32]。常见的TEM型ESBLs还有TEM-10、TEM-12和TEM-26。
2.5.2 SHV型ESBLs (A类) SHV型ESBLs的整体结构与TEM相似[33]。以SHV-1为例,其与TEM-1有68%的同源性,在活性位点附近亦有可变的氨基酸。SHV-5、SHV-12是临床中最常见的SHV型ESBLs。
2.5.3 CTX-M型ESBLs(A类) 一 些 常 见 的A类ESBLs,既不属于SHV型,又不属于TEM型,研究人员将其统称为CTX-M型,意为“善于水解头孢噻肟(cefotaxime)”。现已确证有40余种CTX-M型ESBLs[34],其中传播最为广泛的有CTX-M-14、CTX-M-3、CTX-M-2等。
2.5.4 OXA型ESBLs(D类) OXA型ESBLs得名于“水解苯甲异唑青霉素(oxacillin)”。该类β-内酰胺酶中主要成员有 OXA-1、OXA-2、OXA-10,通过氨基酸替换突变,现已确证了20种OXA。大多数OXA型ESBLs可引起细菌对头孢他啶耐药,此外还可引起克拉维酸耐药。
2.5.5 质粒介导的AmpC(C类) AmpC属于C类β-内酰胺酶,起源并编码于数种革兰阴性菌染色体中。然而随着微生物相互“交流”,目前已有20余种AmpC被发现存在于细菌质粒中[35]。AmpC可引起头霉素、氧基亚氨基β-内酰胺抗生素和克拉维酸的耐药。
2.5.6 碳青霉烯酶(A、B、D类) 碳青霉烯酶(carbapenemases)包括IMP、VIM、KPC几类,它们可引起氧基亚氨基头孢菌素(oxyimino-cephalosporins)和头霉素耐药,且耐受碳青霉烯类抗生素[36]。IMP有17种,得名于该类酶耐受亚胺培南(imipenem)。VIM(Verona integron-encoded metallo-β-lactamase)属于B类,现发现有10种。KPC 则是Klebsiella pneumoniae carbapenemase的缩写,属于A类,由质粒编码。
2.6 蛋白激酶C
蛋白激酶C(protein kinase C,PKC)为丝氨酸/苏氨酸激酶,参与细胞内多条信号通路。基于它们的结构和活化剂不同,蛋白激酶C可分为3个亚族:经典PKCs、新型PKCs和非典型PKCs。其表达水平与ABC家族的表达及细胞凋亡水平相关,可使细菌产生多药耐药性。
经 典PKCs包 括PKCα、PKCβ Ⅰ、PKCβ Ⅱ、PKCγ,具有钙依赖性,并由第二信使——二酰基甘油(DAG)所激活。新型PKCs是非钙依赖的,仍由DAG所激活,包括PKCδ、PKCε、PKCη、PKCθ。而非典型PKCs既不是钙依赖的,也不由DAG作为最佳激动剂,而是由脂质组分(如磷脂酰肌醇、磷脂酸、花生四烯酸和神经酰胺)激活。非典型PKCs包括PKCι、PKCτ和PKCζ等。
3种亚族的性质差异由其结构特点所决定。3种亚族均由C端底物结合催化结构域与N端PS结构域组成。经典PKCs具有钙离子结合结构域C2,这是其钙依赖特点的来源,也是经典PKCs与新型PKCs的唯一区别。非典型PKCs保守DAG结合结构域C1不完整,故而没有DAG激活的能力。非典型PKCs的PB1结构域负责识别PAR-6、 ZIP/p62和丝裂原蛋白激酶5(MEK5)的OPCA基序,NLS和NES分别是核定位序列与核外排序列,均与PKCs 功能相关。与耐药性相关的PKCs主要是PKCα、PKCζ和PKCδ。
2.6.1 PKCα PKCα通过3个方面来增强细胞的耐药性:减少由Gαq介导、磷脂酶C(PLC)-β催化生成的活性氧自由基(ROS);通过磷酸化反应调节抗凋亡蛋白与促凋亡蛋白的表达;抑制PARP的切割活性以对抗凋亡[37]。PKCα还可调节MRP2活性:外排泵MRP2/ ABCC2定位到细胞膜极性侧才能发挥其外排活性,而负责连接细胞膜与细胞骨架的埃兹/根/膜突(ezrin/ radixin/moesin,ERM)蛋白家族中根蛋白(radixin)的C端磷酸化与MRP2定位过程相关,PKCα则参与根蛋白的磷酸化过程。在该通路中PKCα的活性又可由叔丁基氢过氧化物(t-BHP)诱导的GSH含量下降所激活[38](见图4)。而用谷胱甘肽乙酯(GSH-EE)诱导GSH含量恢复,通过PKA/cAMP通路使根蛋白磷酸化,进而使MRP2定位到细胞膜上。
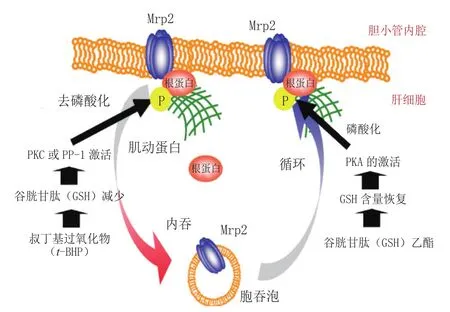
图4 PKCα引起的多药耐药机制Figure 4 Multidrug resistance mechanism caused by PKCα
类似于上述机制,在伤寒沙门氏菌感染的消化道上皮细胞,病原菌感染的信号通过PKAα使埃兹蛋白567位苏氨酸磷酸化,协同MRP2定位在细胞膜极性侧,完成将趋化因子HXA3外排引导多形核白细胞(PMN)迁移至患处的功能。通过以上机制可知,PKCα在GSH-PKC/PKA-ERM-MRP2信号通路发挥重要作用,使细胞产生多药耐药性[39]。
2.6.2 PKCζ PKCζ定位于细胞膜且会从细胞核中外排,但在氧化应激状态下,如细菌感染或吸烟可导致ROS积累,PKCζ会被重分布到细胞核,重新分布到细胞核的PKCζ通过多条与维持细胞增殖相关的信号通路,促使细胞摆脱增殖抑制因子的抑制作用,调整细胞能量代谢结构,使细胞利于转移,协助完成对细胞凋亡的耐受性,从而获得多药耐药性(见图5)[40]。
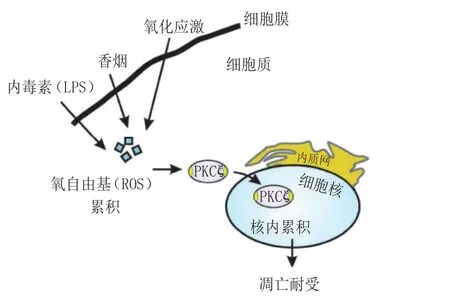
图5 PKCζ耐药性机制Figure 5 Drug resistance mechanism of PKCζ
2.6.3 PKCδ Wnt/β-catenin通路中的G蛋白偶联受体Frizzled 1(FZD 1)激活后上调PKCδ,PKCδ又会激活AP-1活性,AP-1激活其下游基因c-JUN和c-FOS,从更长远的效应来说,又会进一步激活肝细胞生长因子(HGF)与早期生长反应基因1(EGR1),EGR1作为转录因子会影响ABCB1的表达(见图6),从而促使细胞产生耐药性。另外,HGF/EGR1的自分泌效应会对抗细胞的自噬以及凋亡,增加肿瘤细胞的存活能力及耐药能力。总之,PKCδ通过调节ABCB1的表达及对抗凋亡从而导致细胞多药耐药[41]。
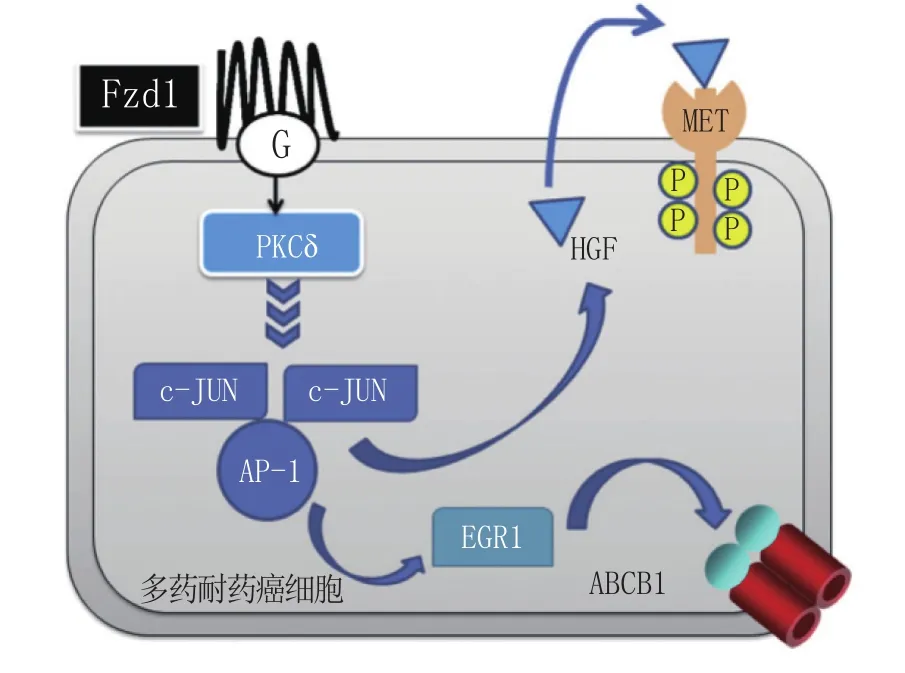
图6 PKCδ引发多药耐药的机制Figure 6 Multidrug resistance mechanism caused by PKCδ
PKC相关的抑制剂已有数个处于临床前研究阶段:Rottlerin作为PKCδ的特异性抑制剂,可以诱发肿瘤细胞凋亡;Gö6976作为PKCα与β Ⅰ的抑制剂,可诱发癌细胞凋亡,同时减少ABCB1在结肠癌细胞中的表达;Ro-318220作为一种广泛的PKC抑制剂可以抑制肿瘤细胞生长。另外,汤森路透Integrity数据库中提到了2013年上市的头孢曲松/舒巴坦的复方制剂,推测其部分机制可能是作为PKC抑制剂来治疗多重耐药的革兰阴性菌感染。
目前,PKC依然是研究较多的靶点,具有很大的开发潜力。机制研究表明,PKC主要从2个方面影响多药耐药性:一是通过调节下游ABC家族转运蛋白的表达调节外排作用,二是通过对细胞凋亡信号通路的影响(ROS的清除及相关蛋白表达的调控)调节细胞凋亡能力。总体来说,针对PKC的活性化合物,尽管发现时间早、研究时间长,但研发难度较大。
3 核糖体
核糖体是蛋白质基因翻译的重要场所,因而对病原微生物和细胞的存活有重要意义。原核生物核糖体由2个核糖核蛋白亚基50S和30S组成,与真核生物核糖体有差异。相应的蛋白质合成抑制剂类抗生素也分为2个子类:30S亚基抑制剂和50S亚基抑制剂。
3.1 30S核糖体亚基
30S核糖体抑制剂包括四环素类抗生素和氨基环醇两类。四环素类抗生素通过阻止氨酰-tRNA与核糖体受体A位点结合而起效,氨基环醇包括大观霉素和氨基糖苷类(如链霉素、卡那霉素、庆大霉素)两类抗生素。它们与30S核糖体亚基中的16S核糖体RNA结合。大观霉素通过抑制延伸因子催化的移位反应干扰肽酰tRNA与核糖体结合稳定性,但不导致蛋白质错译。相比之下,氨基糖苷类与30S核糖体中的16S rRNA的相互作用则能导致mRNA密码子与其同源电荷酰胺-tRNA复合物在核糖体A位的构象发生改变,从而引起tRNA错配和蛋白错译。
7种tet和1种otr环质粒基因的耐药性机制是通过产生核糖体保护蛋白,使核糖体免受四环素类抗生素作用。核糖体保护蛋白为延长因子EF-Tu和EF-G的同源蛋白,其同源性在包含GTP结合区域的N末端尤为显著。Tet(M)和Tet(O)是机制较为清楚的两类核糖体保护蛋白。实验证实,两者依赖核糖体的GTP激酶活性。由于Tet (M)蛋白和EF-G有重叠结合位点,两者可竞争性结合核糖体。Tet (M)蛋白并不能取代EF-Tu及EF-G起延长肽链作用。因Tet (M)蛋白与核糖体亲和力更强,其先与核糖体结合,之后被释放并允许EF-G结合。当Tet (M)、Tet (O)蛋白和GTP同时存在时,四环素类抗生素与30S亚基结合能力下降并从核糖体释放。GTP在GTP激酶作用下水解释放能量导致核糖体构象改变,这一改变不影响正常蛋白质翻译过程,却阻止四环素与30S核糖体结合[42]。
氨基糖苷类抗生素是放线菌的代谢产物。放线菌对自身产生的氨基糖苷类抗生素具有固有耐药性。许多情况下这一耐药性的产生与核糖体保护机制有关。16S rRNA A位特定核苷酸甲基化将会阻碍氨基糖苷类抗生素与30S核糖体亚基结合,从而起到自身保护作用。现已确定16S rRNA的2个位点的甲基化导致2种不同的氨基糖苷类抗生素耐药表型。其中一类16S rRNA甲基化酶对A1408进行甲基化,另一类则对G1405甲基化。前一类对卡那霉素和安普霉素产生耐药性,但对庆大霉素敏感,后一类对卡那霉素和庆大霉素耐药,对安普霉素敏感。这2个核苷酸均位于16S rRNA的A位区域,而氨基糖苷类抗生素正是通过结合这一位点阻碍肽酰-tRNA由A位移位至P位点,从而干扰蛋白正确翻译。
3.2 50S核糖体亚基
50S核糖体抑制剂包括大环内酯类、林可酰胺类、链阳性菌素、氨酰醇以及唑烷酮。50S核糖体抑制剂通过物理性阻碍蛋白质翻译起始过程或肽酰tRNA移位过程抑制肽基转移酶延长新合成多肽链的反应。耐药机制为3个起始因子、30S小亚基与其P位上携带的起始tRNA甲硫氨酸会先于50S核糖体结合在mRNA上。正常情况下,由于唑烷酮类抗生素占据50S核糖体的P位点,50S核糖体因此不能与30S核糖体结合生成70S成熟核糖体,翻译起始过程被终止。若50S已与30S核糖体生成成熟核糖体,则唑烷酮占据50S核糖体P位点可导致A位上氨基酸-tRNA无法转移至P位,翻译过程终止。
50S核糖体突变多为23S rRNA 2 576位的G突变为U,这一结构位于P位点附近。因此可以确定P位点结构突变导致唑烷酮无法再发挥药效。2 032和2 447位的突变则与降低唑烷酮与核糖体结合有关[43]。
大环内酯类、林可酰胺类抗生素和链阳性菌素B的耐药性,也就是所谓的MLSB耐药性表型是由核糖体甲基化引起的。MLSB表型的erm基因编码红霉素核糖体甲基化酶,从而使细菌产生耐药性。在致病菌中,erm蛋白对新生成23S rRNA的单个腺嘌呤进行双甲基化。而位于23S rRNA保守区域的A2058(腺嘌呤)却对以上3种抗生素结合起重要作用。由于甲基化作用,抗生素与靶点的结合被阻碍。大量对大环内酯类、林可酰胺类抗生素产生耐药性的微生物,包括革兰阳性菌、螺旋体以及厌氧菌均表达erm甲基化酶。
至今已有将近40种erm基因被报道。在致病菌中,其多数来源于可自转移的质粒和转座子中。4类erm基因包括:erm(A)、erm(B)、erm(C)和erm(F)。erm(A)和erm(C)存在于金黄色葡萄球菌中,erm(B)大多分布于链球菌和肠球菌,erm(F)则分布于拟杆菌及其他厌氧菌中[44]。
4 相关药物研究
参考汤森路透Intergrity数据库及相关文献,总结了目前具有抗多药耐药作用的药物及活性化合物的研究现状(见表1)。
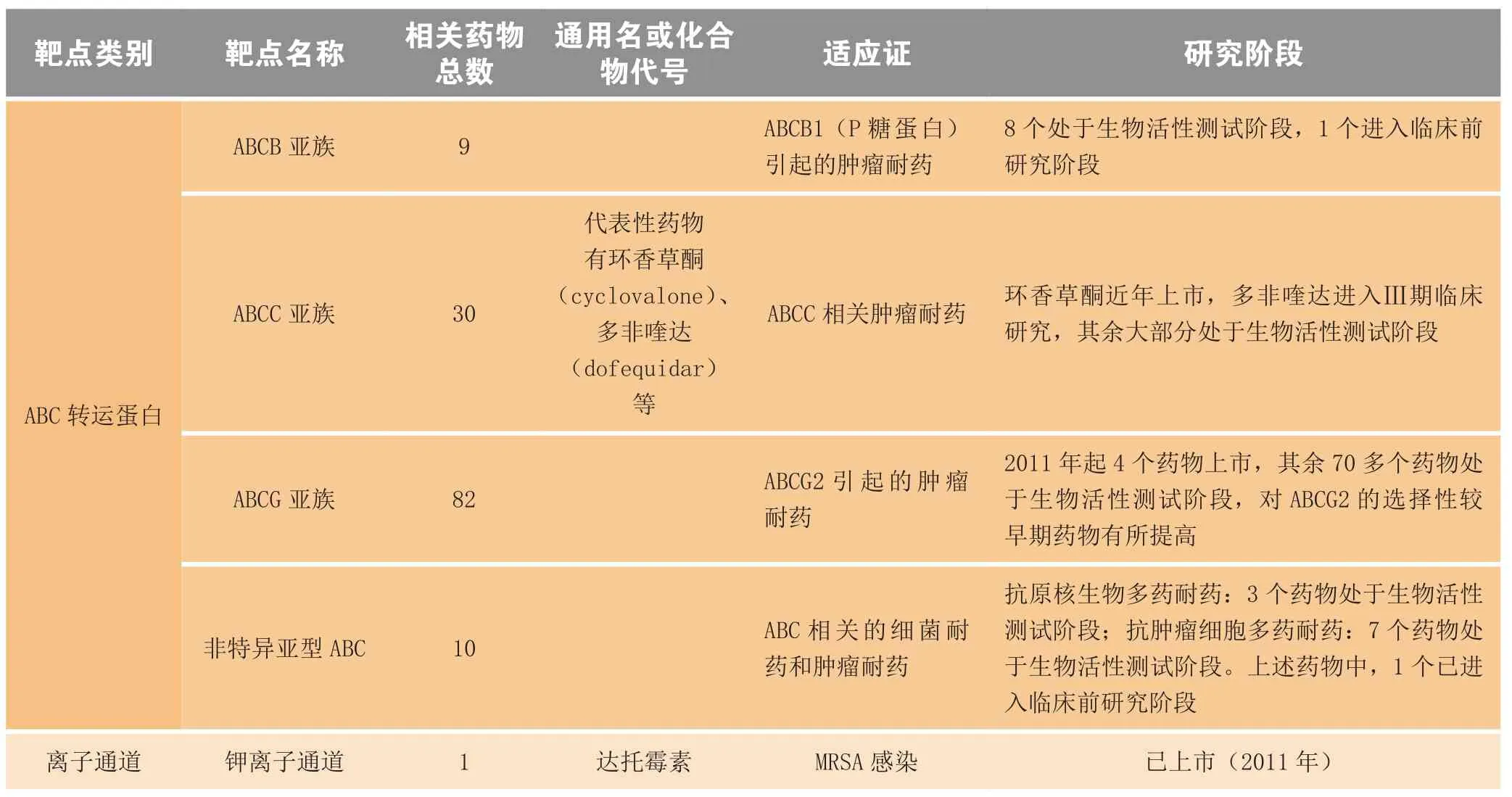
表1 具有抗耐药微生物或肿瘤活性的在研药物Table 1 New drugs in development with activity against resistant microorganisms or tumors

续表1
5 结语
临床上的多药耐药性问题日趋凸显。通过总结多药耐药相关靶点信息和活性化合物的研究状况不难发现,新型的抗多药耐药药物的开发面临着一些困难和挑战,例如,多药耐药的机制相对复杂,编码相关蛋白的基因的突变存在多样性和易变性,还有待深入研究;目前新发现的活性化合物普遍存在溶解性和透膜性较差等问题,需进行结构优化。总之,抗多药耐药药物的研发工作任重而道远。针对目前肿瘤和细菌多药耐药的现状,我们建议从以下角度来进行相关药物的研发:第一,开发新靶点新机制的药物,新的靶点和机制对于解决目前的耐药情况可能是一条便捷、有前景的道路;第二,开发多靶点的药物,多靶点药物可以增强当前使用的药物的治疗效果,缓解耐药性;第三,药物的开发应综合考虑药物代谢动力学性质,以增加临床成功率。
[参 考 文 献]
[1]Szakács G, Paterson J K, Ludwig J A, et al. Targeting multidrug resistance in cancer [J]. Nat Rev Drug Discov,2006,5(3):219-234.
[2]Higgins C F. ABC transporters: from microorganisms to man [J]. Annu Rev Cell Biol,1992,8:67-113.
[3]Chang G, Roth C B. Structure of MsbA from E. coli: a homolog of the multidrug resistance ATP binding cassette (ABC) transporters [J]. Science,2001,293(5536):1793-1800.
[4]Dean M, Rzhetsky A, Allikmets R. The human ATP-binding cassette (ABC) transporter superfamily [J]. Genome Res,2001,11(7):1156-1166.
[5]Tsuruo T, Iida H, Tsukagoshi S, et al. Overcoming of vincristine resistance in P388 leukemia in vivo and in vitro through enhanced cytotoxicity of vincristine and vinblastine by verapamil [J]. Cancer Res,1981,41(5):1967-1972.
[6]Childs S, Yeh R L, Georges E, et al. Identification of a sister gene to P-glycoprotein [J]. Cancer Res,1995,55(10):2029-2034.
[7]Gerloff T, Stieger B, Hagenbuch B, et al. The sister of P-glycoprotein represents the canalicular bile salt export pump of mammalian liver [J]. J Biol Chem,1998,273(16):10046-10050.
[8]Ruetz S, Gros P. Phosphatidylcholine translocase: a physiological role for the mdr2 gene [J]. Cell,1994,77(7):1071-1081.
[9]Van Helvoort A, Smith A J, Sprong H, et al. MDR1 P-glycoprotein is a lipid translocase of broad specificity, while MDR3 P-glycoprotein specifically translocates phosphatidylcholine [J]. Cell,1996,87(3):507-517.
[10]Childs S, Yeh R L, Hui D, et al. Taxol resistance mediated by transfection of the liver-specific sister gene of P-glycoprotein [J]. Cancer Res,1998,58(18):4160-4167.
[11]Smith A J, van Helvoort A, van Meer G, et al. MDR3 P-glycoprotein, a phosphatidylcholine translocase, transports several cytotoxic drugs and directly interacts with drugs as judged by interference with nucleotide trapping [J]. J Biol Chem,2000,275(31):23530-23539.
[12]Dietrich C G, de Waart D R, Ottenhoff R, et al. Mrp2-deficiency in the rat impairs biliary and intestinal excretion and influences metabolism and disposition of the food-derived carcinogen 2-amino-1-methyl-6-phenylimidazo [J]. Carcinogenesis,2001,22(5):805-811.
[13]König J, Rost D, Cui Y, et al. Characterization of the human multidrug resistance protein isoform MRP3 localized to the basolateral hepatocyte membrane [J]. Hepatology,1999,29(4):1156-1163.
[14]Scheffer G L, Kool M, de Haas M, et al. Tissue distribution and induction of human multidrug resistant protein 3[J]. Lab Invest,2002,82(2):193-201.
[15]Belinsky M G, Chen Z S, Shchaveleva I, et al. Characterization of the drug resistance and transport properties of multidrug resistance protein 6(MRP6, ABCC6)[J]. Cancer Res,2002,62(21):6172-6177.
[16]Hopper-Borge E, Chen Z S, Shchaveleva I, et al. Analysis of the drug resistance profile of multidrug resistance protein 7 (ABCC10): resistance to docetaxel [J]. Cancer Res,2004,64(14):4927-4930.
[17]Abbott B L. ABCG2(BCRP) expression in normal and malignant hematopoietic cells [J]. Hematol Oncol,2003,21(3):115-130.
[18]Schinkel A H, Jonker J W. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: an overview [J]. Adv Drug Deliv Rev,2003,55(1):3-29.
[19]Zhao R, Goldman I D. Resistance to antifolates [J]. Oncogene,2003,22(47):7431-7457.
[20]Ozvegy-Laczka C, Cserepes J, Elkind N B, et al. Tyrosine kinase inhibitor resistance in cancer: role of ABC multidrug transporters [J]. Drug Resist Updat,2005,8(1/2):15-26.
[21]Szakács G, Annereau J P, Lababidi S, et al. Predicting drug sensitivity and resistance: profiling ABC transporter genes in cancer cells [J]. Cancer Cell,2004,6(2):129-137.
[22]Mijatovic T, Dufrasne F, Kiss R. Cardiotonic steroids-mediated targeting of the Na(+)/K(+)-ATPase to combat chemoresistant cancers [J]. Curr Med Chem,2012,19(5):627-646.
[23]Amaral L, Martins M, Viveiros M. Enhanced killing of intracellular multidrug-resistant Mycobacterium tuberculosis by compounds that affect the activity of efflux pumps [J]. J Antimicrob Chemother,2007,59(6):1237-1246.
[24]Alekshun M N, Levy S B. Levy.Molecular mechanisms of antibacterial multidrug resistance[J]. Cell,2007,128(6):1037-1050.
[25]Yoshida H, Bogaki M, Nakamura M, et al. Quinolone resistancedetermining region in the DNA gyrase gyrB gene of Escherichia coli [J]. Antimicrob Agents Chemother,1991,35(8):1647-1650.
[26]Hooper D C. Mechanisms of fluoroquinolone resistance [J]. Drug Resist Updat,1999,2(1):38-55.
[27]Kohanski M A, Dwyer D J, Collins J J. How antibiotics kill bacteria: from targets to networks [J]. Nat Rev Microbiol,2010,8(6):423-435.
[28]Sköld O. Sulfonamide resistance: mechanisms and trends[J]. Drug Resist Updat,2000,3(3):155-160.
[29]Heath R J, Rubin J R, Holland D R, et al. Mechanism of triclosan inhibition of bacterial fatty acid synthesis [J]. J Biol Chem,1999,274(16):11110-11114.
[30]Jacoby G A, Munoz-Price L S. The new β-lactamases[J]. New Engl J Med,2005,352(4):380-391.
[31]Huang W, Petrosino J, Hirsch M, et al. Amino acid sequence determinants of beta-lactamase structure and activity [J]. J Mol Biol,1996,258(4):688-703.
[32]Nukaga M, Mayama K, Hujer A M, et al. Ultrahigh resolution structure of a class A beta-lactamase: on the mechanism and specificity of the extendedspectrum SHV-2 enzyme [J]. J Mol Biol,2003,328(1):289-301.
[33]Kuzin A P, Nukaga M, Nukaga Y, et al.Structure of the SHV-1β-lactamase [J]. Biochemistry,1999,38(18):5720-5727.
[34]Bonnet R. Growing group of extended-spectrum beta-lactamases: the CTX-M enzymes [J]. Antimicrob Agents Chemother,2004,48(1):1-14.
[35]Philippon A, Arlet G, Jacoby G A. Plasmid-determined AmpC-type betalactamases [J]. Antimicrob Agents Chemother,2002,46(1):1-11.
[36]Nordmann P, Poirel L. Emerging carbapenemases in Gram-negative aerobes [J]. Clin Microbiol Infect,2002,8(6):321-331.
[37]Lee S K, Shehzad A, Jung J C, et al. Protein kinase Cα protects against multidrug resistance in human colon cancer cells [J]. Mol Cells,2012,34(1):61-69.
[38]Sekine S, Ito K, Saeki J, et al. Interaction of Mrp2 with radixin causes reversible canalicular Mrp2 localization induced by intracellular redox status [J]. Biochim Biophys Acta,2011,1812(11):1427-1434.
[39]Agbor T A, Demma Z C, Mumy K L, et al. The ERM protein, ezrin, regulates neutrophil transmigration by modulating the apical localization of MRP2 in response to the SipA effector protein during Salmonella Typhimurium infection [J]. Cell Microbiol,2011,13(12):2007-2021.
[40]Rimessi A, Patergnani S, Ioannidi E, et al. Chemoresistance and cancerrelated inflammation: two hallmarks of cancer connected by an atypical link, PKCζ [J]. Front Oncol,2013,3:232.
[41]Hung T H, Chen C M, Tseng C P, et al. FZD1 activates protein kinase C delta-mediated drug-resistance in multidrug-resistant MES-SA/Dx5 cancer cells [J]. Int J Biochem Cell Biol,2014,53:55-65.
[42]Chopra I, Roberts M. Tetracycline antibiotics: mode of action, applications, molecular biology, and epidemiology of bacterial resistance [J]. Microbiol Mol Biol Rev,2001,65(2):232-260.
[43]Bozdogan B, Appelbaum P C. Oxazolidinones: activity, mode of action, and mechanism of resistance [J]. Int J Antimicrob Agents,2004,23(2):113-119.
[44]Leclercq R. Mechanisms of resistance to macrolides and lincosamides: nature of the resistance elements and their clinical implications [J]. Clin Infect Dis,2002,34(4):482-492.
Research Progress in Therapeutic Targets for Multidrug Resistance and Related Drugs
WU Chen, XU Zhuxuan, ZHOU Qimeng, ZHI Yunbao, LIN Kejiang
( Department of Pharmacy, China Pharmaceutical University, Nanjing 210009, China)
Multidrug resistance (MDR) refers to the process in which cells produce wide resistance to a variety of drugs, leading to unsatisfied treatment. Multidrug resistance mainly occurs in treatment of infections and cancers, which greatly restricts the therapeutic effects on both diseases. The research progress of anti-MDR mechanisms and drugs based on the receptor types, including transport proteins, ion channels, enzymes and ribosomes, has been reviewed in this paper, so as to provide references for the development of anti-MDR drugs.
multidrug resistance; infection; tumor; transporter; enzyme; ribosome protein
R966
A
1001-5094(2014)11-0829-12
接受日期:2014-09-22
*通讯作者:林克江,副教授;
研究方向:新药设计;
Tel:025-83271351; E-mail:link@cpu.edu.cn
